

Abstract
Modification of natural products with prenyl groups and the ensuing oxidative transformations are important for introducing structural complexity and biological activities. Understanding the different mechanisms Nature performs prenylation can lead to new enzymatic tools. Penigequinolones (1) are potent insecticidal alkaloids that contain a highly modified ten-carbon prenyl group. Here we reveal an iterative prenylation mechanism for installing the ten-carbon unit using two aromatic prenyltransferases (PenI and PenG) present in the gene cluster of 1 from Penicillium thymicola. The initial Friedel-Crafts alkylation is catalyzed by PenI to yield the dimethylallyl quinolone 6. The five-carbon side chain is then dehydrogenated by a Flavin-dependent monooxygenase to an aryldiene 9, which serves as the electron-rich substrate for a second alkylation with dimethylallyl diphosphate to yield a stryrenyl product 10. The completed, oxidized ten-carbon prenyl group is then shown to undergo further structural morphing to yield yaequinolone C 12, the immediate precursor of 1. Our studies therefore uncover an unprecedented prenyl chain extension mechanism in natural product biosynthesis.
An important biosynthetic transformation that generates chemical diversity in natural products is the addition of prenyl groups.1–4 The transfer of prenyl groups, together with the subsequent modifications, significantly expand structural complexity and biological activities of all major families of natural products, including polyketides,5–6 nonribosomal peptides7–8 and indole alkaloids,9–12 etc. The different prenyl precursors that are used to decorate natural products typically include dimethylallyl diphosphate (DMAPP), geranyl diphosphate (GPP), farnesyl diphospate (FPP) or geranylgeranyl diphosphate (GGPP).13 Formation of these building blocks are catalyzed by isoprenyl diphosphate synthases (IPPSs) through the iterative, head-to-tail addition of isopentenyl diphosphate (IPP) extender units.14 Prenyl groups of the desired size are then transferred to an electron-rich substrate by a family of enzymes collectively known as prenyltransferases (PTases).15 Notwithstanding this canonical model of preassembly followed by transfer of intact prenyl groups, a number of bioactive natural products contain prenyl substructures may be synthesized by unusual mechanisms. Examples include the twenty-five carbon lipid portion found in moenomycin,16 the seven-carbon side chain found in mycophenolic acid,17 the ten-carbon cyclolavandulyl skeleton found in lavanducyanin,18 and the styrenyl-like framework found in fungal quinolone alkaloids represented by penigequinolone (1) as shown in Figure 1. Understanding new mechanisms of prenyl group transfer and modification will therefore lead to the expansion of enzymatic tools that can modify natural products with prenyl functionalities.
Figure 1.

Representative prenylated natural products with unusual prenyl group arrangements. a. Proposed biosynthetic pathway of penigequinolones family compounds. The proposed geranyl isoprenoid unit is shown in red. b. natural products contains unusually arranged isoprenoid groups.
Penigequinolones produced by various Penicillium sp. and Aspergillus sp. are quinolone alkaloids with oxidized prenyl groups, which are crucial for the potent insecticidal activities.19–26 The core of this family of molecules is the 6,6-bicyclic quinolone 2 that is derived from the oxidative rearrangement of methoxycyclopeptin 7, which is the product of an nonribosomal peptide formed through the condensation between anthranilate and O-methyl-tyrosine.27 C7 of the phenolic ring in 2 is proposed to undergo Friedel-Crafts geranylation to yield the proposed intermediate peniprequinolone 3 (Figure 1a).27 Significant morphing of the geranyl unit is then proposed to take place to yield the various natural products, including penigequinolone A (1a) and B (1b) that contain the gem-dimethyl pyran ring;22,26 aspoquinolone A (4a) and B (4b) that contain a fused cyclopropane-tetrahydrofuran unit;23 and the chromene-containing yaequinolone J1 (5a) and J2 (5b).24
The prenyl transfer and modifications steps that transform 2 are of interest from several perspectives, including i) the quinolone scaffold as a substrate of C-prenyltransfer has not been reported previously, whereas prenylation of L-tyrosine, indole and polycyclic aromatics have been well-documented;4–6,9,11–12,28 ii) all the natural products derived from the proposed intermediate 3 are heavily oxidized, including the double bond at C1′-C2′. The common oxygen atom at C3′ is proposed to derive from the epoxidation of C2′-C3′ and/or C7′-C8′ double bonds in 3 and subsequent ring-opening;23 and iii) formation of the pyran ring in 1 as result of an unusual C-C bond rearrangement. Here we demonstrate 3, which is the product of the proposed geranyltransfer reaction,23,27 is in fact not an intermediate of the biosynthetic pathway. Instead, we show prenylation of the quinolone occurs in a unprecedented, step-wise fashion, in which two DMAPP units are iteratively added to 2 to afford an oxidized ten-carbon isoprenoid unit that is primed for subsequent rearrangement reactions. Such iterative elongation of prenyl groups has not been previously observed in natural product biosynthesis.
To understand the prenylation and subsequent modification steps, we sequenced the genome of Penicillium thymicola IBT5891, a producing strain of 1.29 Using the previously characterized cyclopenase AsqJ that converts 7 to 8 as a lead,30 the putative pen cluster was located and shown in Figure 2a. The gene cluster contains the expected nonribosomal peptide synthetase (NRPS) PenN, the AsqJ homolog PenM, and an assortment of redox and methyltransferase enzymes (Table S2). Unexpectedly, the gene cluster encodes two aromatic prenyltransferases PenG and PenI,31 instead of the single geranyltransferase that was expected to build the proposed intermediate 3. Genetic deletion of penN (Figure S1) using a split-marker approach and hygromycin as the resistance marker led to abolishment of 1 (Figure 2b, i & ii), thereby confirming the pen cluster is responsible for the biosynthesis of 1.
Figure 2.

Confirmation of pen gene cluster in P. thymicola. a Organization and proposed function of the pen gene cluster. b. LC-MS analyses of culture extracts from wild type P. thymicola and mutant strains. Peak labeled by asterisk is fumiquinazoline F, an unrelated metabolite.
We initially reasoned the presence of two prenyltransferases may account for the parallel modification of 2 with GPP that yields 3, and with DMAPP that yields peniprequinolone 6, which was identified from different Penicillium sp.22,25–26 However, no trace of 6 was found in the wild type strain. Bioinformatic and phylogenetic analyses showed that i) PenI is closely related to NscD and VrtD that catalyze the Friedel-Crafts prenylation of polycyclic aromatic polyketides;5–6 and ii) PenG clades independently from other fungal prenyltransferases and is more closely related to dimethylallyltryptophan synthases (DMATS) (Figure S2).11–12,32 To investigate the roles of PenG and PenI, both enzymes were cloned from cDNA, expressed and purified from Escherichia coli to homogeneity (Figure S3). We then constructed a knockout cassette that targeted the entire region that included penG-penI. The mutant strain was unable to produce 1 and accumulated 2 (Figure 2b, iii), which was purified and used as substrate for biochemical assays (Table S3).
Compound 2 was first incubated with 2 mM GPP, MgCl2, and either PenG or PenI. Unexpectedly, neither enzyme converted 2 to the proposed intermediate 3 (Figure 3, i & ii). In contrast, when DMAPP was used as the substrate, PenI catalyzed the near complete conversion of 2 into a new compound with mass consistent with the addition of one five-carbon unit and that of 6 (m/z 382 [M-H]−), while PenG remained inactive (Figure 3, iii & iv). To elucidate the structure of the product, a whole-cell biotransformation using E. coli expressing PenI was performed followed by product extraction and purification. Complete 1H and 13C characterization confirmed the identity of the compound as 6 (Figure 3 and Table S4).26 The in vitro results show that neither prenyltransferases in the pen cluster can function as a geranyltransferase using 2 as a substrate. Instead, PenI is a dimethylallyl transferase, while the function of PenG remained unresolved. However, these results point to the possibility that 6 is an intermediate in the biosynthetic pathway of 1 instead of 3. Indeed, feeding 6 into the ΔpenN blocked mutant of P. thymicola restored the production of 1 (Figure 2b, iv), confirming the existence of an prenyl elongation mechanism downstream in the pathway.
Figure 3.
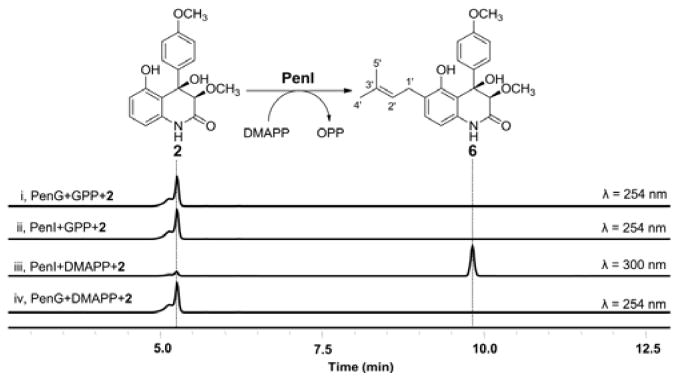
In vitro biochemical analyses of PenG and PenI with 2. All reactions performed with purified PTases in the presence of MgCl2 and the substrate 2.
The head-to-tail polyprenyl elongation catalyzed by IPPS requires the terminal Δ3 double bond present in IPP.13 Therefore compound 6, which contains the internal Δ2 double bond, cannot directly attack a dimethylallyl cation intermediate to yield a ten-carbon prenyl group. With the hypothesis that PenG catalyzes the prenyl elongation step, we constructed the ΔpenG mutant in order to identify a possible electron-rich substrate. LC-MS analysis of the extract showed the loss of 1 and the appearance of a new compound 9 (m/z 380 [M-H]−) that has a red-shifted λmax compared to 6 (from 300 nm to 337 nm) (Figure 4a, i & ii). Isolation and structural characterization of 9 revealed the compound to be the aryldiene quinolone previously isolated as yaequinolone E (Figure 4 and Table S4).22 Compared to 6, 9 has undergone one dehydrogenation followed by shifting of the Δ2 double bond to the Δ3′ position. To confirm that 9 is an authentic intermediate of 1, feeding of 9 to the ΔpenN mutant strain was performed. As expected, restoration of the biosynthesis of 1 was observed (Figure 4a, iii).
Figure 4.

Mapping the prenyl elongation step in biosynthesis of 1. a. LC-MS analyses of culture extracts from P. thymicola strains. b. Analyses of in vitro biochemical assays involving PenG or PenI with 9.
To assay the chain elongation reaction, purified 9 was incubated with PenG, DMAPP and MgCl2 followed by extraction and LC-MS analysis. Near complete conversion of 9 to one major compound 10 (m/z 466 [M-H]−), as well as other two minor compounds (m/z 432 [M+H-H2O]+) were observed (Figure 4b, i). In contrast, no reaction occurred when PenI was used as the prenyltransferase (Figure 4b, ii). To obtain sufficient amounts of 10 and the minor products for structural characterization, a large scale biotransformation using S. cerevisiae expressing PenG was performed. All three compounds were thoroughly characterized by 1D- and 2D-NMR (Table S5–S6), and are confirmed to be products containing ten-carbon prenyl groups. Compound 10 is the styrenyl quinolone containing a C3′-hydroxyl prenyl chain. Feeding of 10 to the ΔpenN block mutant efficiently restored the biosynthesis of 1 (Figure 4, iv), confirming the compound as an on pathway intermediate of the pathway. We reasoned that 10 can be obtained from the addition of H2O to the cationic intermediate 11 following the PenG-catalyzed chain elongation reaction (Figures 4 and 5). This mechanism is verified by performing the PenG reaction in H218O buffer and observing the incorporation of 18O into 10 (Figure S4). The two minor compounds from the in vitro reaction are the yaequinolone J1 and J2 (5a and 5b),24 which are presumably shunt products in the biosynthesis of 1 derived from the cyclization of 11 through the phenolate oxygen (Figure 5). Interestingly, feeding of 10 to ΔpenN led to significant accumulation of 5a and 5b (Figure 4, iv), suggesting that under fungal culturing conditions, the loss of H2O by 10 to yield 11 (reverse reaction to generate the cation) may be significant. In contrast, purified 10 is stable and does not undergo spontaneous conversion to 5a and 5b.
Figure 5.

Updated biosynthetic pathway of 1.
Having established that the ten-carbon isoprenoid side chain in 1 is derived from the iterative addition of two DMAPP molecules, first from 2 to 6, followed by 9 to 10, we next aimed to identify the oxidative enzymes responsible for the conversion of 6 to 9, as well as the downstream modification of 10. The enzyme encoded by penH is a membrane-bound Flavin-dependent monooxygenase (FMO) featuring both BBE33 and GlcD34 conserved domains. GlcD-containing FMOs are membrane-bound respiratory D-lactate dehydrogenase, which catalyze the conversion of D-lactate to pyruvate using a covalently linked FAD as a co-factor.34 Hence, PenH is likely a FAD-dependent dehydrogenase that may catalyze the dehydrogenation of 6 to 9. This may proceed via base-catalyzed removal of C1′ hydrogen and capturing of the C4′ hydrogen as a hydride by the FAD cofactor (Figure 5).35 Indeed, the ΔpenH mutant accumulated 6 as the only product, and the biosynthesis of 1 can be restored through the feeding of 9 (Figure 4a, v & vi). Attempts to express PenH as a soluble protein from either E. coli or S. cerevisiae were not successful, thereby precluding the direct assay of this reaction using purified enzyme. Nevertheless, the identification of the role of PenH using genetic methods allowed us to map the entire pathway that converts the quinolone 2 to the first ten-carbon isoprenyl containing intermediate 10 as shown in Figure 5. The pathway starts with the canonical Friedel-Crafts alkylation of 2 with dimethylallyl cation by PenI to yield 6, which is subjected to FAD-dependent dehydrogenation to yield the conjugated diene 9. The Δ3′ double bond then serves as the site of the second alkylation with DMAPP catalyzed by PenG to yield the carbenium ion intermediate 11, which can be attacked by H2O to yield 10 or undergo cyclization to yield 5a and 5b (Figure 5).
We next targeted the identification of enzymes that modify 10 into yaequinolone C (12) (Figure 5), which is proposed to be the immediate precursor of 1.23 The conversion likely involves epoxidation of the terminal C7′–C8′ olefin in 10 to yield 13, followed by epoxide ring opening initiated by the C3′ hydroxyl group to yield the tetrahydrofuran-containing 12. Based on this hypothesis, we performed genetic knockout of the FMO PenE, which displays sequence homology to PaxM, a FMO catalyzing the epoxidation of the diterpene unit in the biosynthesis of fungal indole-diterpenes.36 Inactivation of PenE led to the disappearance of 1 and the accumulation of 10, as well as the minor metabolites 5a and 5b (Figure 4a, vii). This is consistent with PenE functioning as the epoxidase as shown in Figure 5. To further probe the role of PenE in the proposed morphing of 10, we performed biotransformation experiments using S. cerevisiae as an expression host. When 10 was added to the yeast culture expressing PenE alone, we detected the formation of a new compound that is consistent with the molecular weight of 12 (m/z 482 [M-H]−), as well as the formation of shunt products 5a and 5b (Figure S5, ii). Compound 12 was purified and characterized by NMR to be identical to yaequinolone C (Table S7).22 The successful transformation of 10 to 12 suggests that the epoxide-opening of 13 may take place spontaneously. Interestingly, the yield of 12 was significantly elevated (~10 fold) when the biotransformation was performed in yeast host coexpressing both PenE and PenJ, a predicted cysteine hydrolase (Figure S5, iii). Increase in conversion of 10 to 12 therefore indicates the role of PenJ as an epoxide hydrolase in enhancing the rate of the 5-exo-tet cyclization step.
In conclusion, we identified an unprecedented mechanism of prenyl elongation in the biosynthesis of penigequinolone family of natural products. Two prenyltransferases successively transferred dimethylallyl units to the quinolone core 2 to yield 10 that contains a “pseudo-geranyl” moiety. Activation of the first dimethylallyl unit to the conjugated diene 9 is accomplished by a FAD-dependent dehydrogenation. The hydroxylated isoprenoid unit in 10 is optimally setup to undergo cyclization towards formation of the final natural products. Our work therefore reveals a new strategy employed by nature to transfer and tailor prenyl group to natural products towards generation of structural complexity.
Supplementary Material
Acknowledgments
This work was supported by NIH 1R56AI101141 and 1DP1GM106413 to Y.T. NMR instrumentation was supported by the NSF equipment grant CHE-1048804. Z.Z. and D.L. are supported by fellowship from the China Scholarship Council (CSC).
Footnotes
Experimental details and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Yazaki K, Sasaki K, Tsurumaru Y. Phytochemistry. 2009;70:1739. doi: 10.1016/j.phytochem.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 2.Li SM. Appl Microbiol Biotechnol. 2009;84:631. doi: 10.1007/s00253-009-2128-z. [DOI] [PubMed] [Google Scholar]
- 3.Tello M, Kuzuyama T, Heide L, Noel JP, Richard SB. Cell Mol Life Sci. 2008;65:1459. doi: 10.1007/s00018-008-7579-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuzuyama T, Noel JP, Richard SB. Nature. 2005;435:983. doi: 10.1038/nature03668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chooi YH, Fang J, Liu H, Filler SG, Wang P, Tang Y. Org Lett. 2013;15:780. doi: 10.1021/ol303435y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chooi YH, Wang P, Fang J, Li Y, Wu K, Tang Y. J Am Chem Soc. 2012;134:9428. doi: 10.1021/ja3028636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balibar CJ, Howard-Jones AR, Walsh CT. Nat Chem Biol. 2007;3:584. doi: 10.1038/nchembio.2007.20. [DOI] [PubMed] [Google Scholar]
- 8.Liu X, Walsh CT. Biochemistry. 2009;48:11032. doi: 10.1021/bi901597j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsh CT. ACS Chem Biol. 2014;9:2718. doi: 10.1021/cb500695k. [DOI] [PubMed] [Google Scholar]
- 10.Li SM. Nat Prod Rep. 2010;27:57. doi: 10.1039/b909987p. [DOI] [PubMed] [Google Scholar]
- 11.Li SM. Phytochemistry. 2009;70:1746. doi: 10.1016/j.phytochem.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 12.Steffan N, Grundmann A, Yin WB, Kremer A, Li SM. Curr Med Chem. 2009;16:218. doi: 10.2174/092986709787002772. [DOI] [PubMed] [Google Scholar]
- 13.Dewick PM. Nat Prod Rep. 2002;19:181. doi: 10.1039/b002685i. [DOI] [PubMed] [Google Scholar]
- 14.Kharel Y, Koyama T. Nat Prod Rep. 2003;20:111. doi: 10.1039/b108934j. [DOI] [PubMed] [Google Scholar]
- 15.Liang PH, Ko TP, Wang AH. Eur J Biochem. 2002;269:3339. doi: 10.1046/j.1432-1033.2002.03014.x. [DOI] [PubMed] [Google Scholar]
- 16.Ostash B, Walker S. Nat Prod Rep. 2010;27:1594. doi: 10.1039/c001461n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regueira TB, Kildegaard KR, Hansen BG, Mortensen UH, Hertweck C, Nielsen J. Appl Environ Microbiol. 2011;77:3035. doi: 10.1128/AEM.03015-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozaki T, Zhao P, Shinada T, Nishiyama M, Kuzuyama T. J Am Chem Soc. 2014;136:4837. doi: 10.1021/ja500270m. [DOI] [PubMed] [Google Scholar]
- 19.An CY, Li XM, Luo H, Li CS, Wang MH, Xu GM, Wang BG. J Nat Prod. 2013;76:1896. doi: 10.1021/np4004646. [DOI] [PubMed] [Google Scholar]
- 20.Neff SA, Lee SU, Asami Y, Ahn JS, Oh H, Baltrusaitis J, Gloer JB, Wicklow DT. J Nat Prod. 2012;75:464. doi: 10.1021/np200958r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uchida R, Imasato R, Yamaguchi Y, Masuma R, Shiomi K, Tomoda H, Omura S. J Antibiot (Tokyo) 2006;59:646. doi: 10.1038/ja.2006.86. [DOI] [PubMed] [Google Scholar]
- 22.Uchida R, Imasato R, Tomoda H, Omura S. J Antibiot (Tokyo) 2006;59:652. doi: 10.1038/ja.2006.87. [DOI] [PubMed] [Google Scholar]
- 23.Scherlach K, Hertweck C. Org Biomol Chem. 2006;4:3517. doi: 10.1039/b607011f. [DOI] [PubMed] [Google Scholar]
- 24.Uchida R, Imasato R, Shiomi K, Tomoda H, Omura S. Org Lett. 2005;7:5701. doi: 10.1021/ol052458h. [DOI] [PubMed] [Google Scholar]
- 25.He J, Lion U, Sattler I, Gollmick FA, Grabley S, Cai J, Meiners M, Schunke H, Schaumann K, Dechert U, Krohn M. J Nat Prod. 2005;68:1397. doi: 10.1021/np058018g. [DOI] [PubMed] [Google Scholar]
- 26.Kusano M, Koshino H, Uzawa J, Fujioka S, Kawano T, Kimura Y. Biosci Biotechnol Biochem. 2000;64:2559. doi: 10.1271/bbb.64.2559. [DOI] [PubMed] [Google Scholar]
- 27.Walsh CT, Haynes SW, Ames BD, Gao X, Tang Y. ACS Chem Biol. 2013;8:1366. doi: 10.1021/cb4001684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heide L. Curr Opin Chem Biol. 2009;13:171. doi: 10.1016/j.cbpa.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 29.Frisvad JC, Smedsgaard J, Larsen TO, Samson RA. Stud Mycol. 2004;49:201. [Google Scholar]
- 30.Ishikawa N, Tanaka H, Koyama F, Noguchi H, Wang CC, Hotta K, Watanabe K. Angew Chem Int Ed Engl. 2014;53:12880. doi: 10.1002/anie.201407920. [DOI] [PubMed] [Google Scholar]
- 31.Bonitz T, Alva V, Saleh O, Lupas AN, Heide L. PLoS One. 2011;6:e27336. doi: 10.1371/journal.pone.0027336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Metzger U, Schall C, Zocher G, Unsold I, Stec E, Li SM, Heide L, Stehle T. Proc Natl Acad Sci U S A. 2009;106:14309. doi: 10.1073/pnas.0904897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kutchan TM, Dittrich H. J Biol Chem. 1995;270:24475. doi: 10.1074/jbc.270.41.24475. [DOI] [PubMed] [Google Scholar]
- 34.Reed DW, Hartzell PL. J Bacteriol. 1999;181:7580. doi: 10.1128/jb.181.24.7580-7587.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh CT, Wencewicz TA. Nat Prod Rep. 2013;30:175. doi: 10.1039/c2np20069d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tagami K, Liu C, Minami A, Noike M, Isaka T, Fueki S, Shichijo Y, Toshima H, Gomi K, Dairi T, Oikawa H. J Am Chem Soc. 2013;135:1260. doi: 10.1021/ja3116636. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.