

Abstract
Systemic lupus erythematosus (SLE) is a complex multisystem autoimmune disease, characterized by a spectrum of autoantibodies that target multiple cellular components. Glomerulonephritis is a major cause of morbidity in patients with SLE. Little is known about the pathogenesis of SLE renal damage and compromised renal function. Activation of both Stat1 and Stat3 has been reported in lupus and lupus nephritis. The reciprocal activation of these two transcription factors may have a major impact on renal inflammation. To study the role of Stat1 in a lupus model, we induced lupus-like chronic graft-versus-host disease (cGVHD) in Stat1-KO and in WT mice by intraperitoneal injection of class II-disparate bm12 splenocytes. WT recipients of these alloreactive cells developed anti-dsDNA autoantibodies starting at week 2 as expected, with a decline after week 4. In contrast, Stat1-KO hosts exhibited a prolonged and significant increase of anti-dsDNA autoantibody responses compared to WT mice (week 4 to week 8). Increased autoantibody titers were accompanied by increased proteinuria and mortality in the cGVHD host mice lacking Stat1. Further analysis revealed expression and activation of Stat3 in the glomeruli of Stat1-KO host mice but not WT mice with cGVHD. Glomerular Stat3 activity in the Stat1-KO mice was associated with increased IL-6 and IFN-γ secretion and macrophage infiltration. Interactions between Stat1 and Stat3 thus appear to be crucial in determining the severity of lupus-like disease in the cGVHD model.
Keywords: Stat1, Stat3, lupus nephritis, cGVHD
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disorder associated with immune-complex mediated multiple-organ damage (1, 2). A major contributor to morbidity and mortality among patients with SLE is renal inflammation, known as lupus nephritis (LN) (3). The failures in immunoregulation of lupus are still poorly understood (1, 4).
Interferons and interferon-inducible genes are thought to be important in the pathogenesis of SLE. The “interferon signature”, consisting of increased transcription of both type I interferon and of type I interferon-activated genes, is frequently observed in cells from SLE patients and is associated with disease activity. Because type I interferons (IFN-I) activate Stat1, Stat2, and Stat3, these transcription factors may have key roles in SLE by either promoting or suppressing inflammation (5). Type II interferon (IFN-γ) can also activate Stat1 and Stat3 (6). Stat1 is present in renal biopsies of lupus nephritis patients and kidney sections of lupus-prone mice, and expression levels correlate with disease activity (7). Stat3 activation has also been identified in the normal human kidney, and markedly increased expression of Stat3 is seen in many forms of glomerulonephritis (8). Stat1 and Stat3 play opposing roles in a variety of cell functions (9-11). In mouse embryonic fibroblasts, IFN-γ weakly activates Stat3, but in the absence of Stat1, Stat3 activation is increased and prolonged (6). IL-6 can activate Stat3 through the gp130 subunit and mediate the diverse effects of Stat3 in development and diseases (12, 13). Gain-of-function Stat3 mutations lead to early-onset autoimmune disease in patients, with clinical improvement observed after IL-6 blockade (14). These many observations led us to consider investigation in depth of the roles of Stats in a lupus animal model.
Bm12-induced chronic graft-versus-host disease (cGVHD) is initiated with injection of MHC-II incompatible spleen cells and results in a chronic syndrome characterized by the production of high titers of a spectrum of autoantibodies similar to those seen in human SLE, accompanied by some of the pathological manifestations of SLE. The autoantibodies produced in the syndrome come entirely from the recipient B cells (15). The pattern of autoantibody production and the renal pathology produced in the murine cGVH model are quite consistent with SLE. The disease progresses over a period of weeks, with substantial mortality in the recipient animals. Antibody titers then regress in surviving animals. This model has been demonstrated to be valuable for elucidating the fundamental immunopathophysiology of SLE (4, 15). In the present report, we explored the role of Stat1 and Stat3 in renal inflammation in mice undergoing cGVHD. We were surprised to find prolonged and markedly elevated autoantibody responses in mice lacking Stat1. Also unanticipated was the finding that of Stat3 protein levels and its activation were substantially increased in Stat1-KO mice subjected to cGVHD. This Stat3 activation was associated with IL-6 and IFN-γ expression and with glomerular macrophage infiltration. These studies underscore the complexity of Stat dysregulation in SLE renal disease, but also open possibilities for the therapeutic utility of selective modulators of Stat1/Stat3 signaling in the management of renal inflammation in SLE patients.
Materials and Methods
Mice
C57BL/6 (WT, B6: H-2b, Ighb), B6.Stat1-KO, B6.Stat2-KO, and B6.C-H2bm12 (bm12: H-2bm12, Ighb) mice were maintained on a 24-h light-dark cycle, with food and water available ad libitum. All mice in the study were on C57BL/6 background male mice (>10-generation backcrossed, non-littermate) between 8-12 weeks of age. All experimental animal procedures were approved by and in accordance with the Institutional Animal Care and Use Committee of Temple University.
Induction of cGVHD
Chronic GVHD was induced as previously described (16). Briefly, B6.WT or B6.Stat1-KO recipient mice were injected intraperitoneally with 100 million splenocytes from bm12 mice. Serum samples were prepared from peripheral blood on the day of injection and at weekly intervals. Mouse sera were stored at −40°C for autoantibody analysis.
Kidney function and histological analysis
Chronic GVHD mice were followed weekly with measurement of proteinuria (Uristix, Bayer Corporation, Elkhart, IN). Kidneys were removed at time points indicated at figure legends and frozen in OCT medium. 4 μm sections were stained with H&E. Pathological evaluation by light microscopy was done in a blinded manner by Dr. Mark Birkenbach.
Flow Cytometry
Mice were sacrificed and spleens were harvested in cold RPMI-1640 medium. Single-cell suspensions were obtained after red blood cell lysis. Kidneys were also harvested, cut into <1mm pieces, and subjected to collagenase I and DNase digestion. Single-cell suspensions were obtained after EDTA incubation (17). Staining was done in staining buffer with various combination of antibodies at 4°C for 40 minutes. Cells were then washed and data were acquired using a FACS Canto flow cytometer (BD Immunocytometry Systems, San Jose, CA). Data were analyzed with FlowJo software (San Carlos, CA).
Autoantibody ELISA
Anti-dsDNA, anti-chromatin, and autoantibody isotypes were quantitated as published (18). Briefly, 96-well plates were coated with either dsDNA (2.5 μg/ml) or chromatin (3 μg/ml). Poly L-lysine (1 μg/ml, Sigma-Aldrich, St. Louis, MO) pre-treatment was required for the dsDNA plate. Serum samples were diluted in borate-buffered saline and incubated overnight at 4°C. Alkaline phosphatase-conjugated goat anti-mouse total IgG or IgG isotypes (Jackson ImmunoResearch Laboratories, West Grove, PA) were added to detect the autoantibody levels. The plates were washed and incubated with substrate at 37°C and read at various time points with an ELISA reader at 405nm.
Immunofluorescent staining
Mouse kidneys were snap-frozen in liquid nitrogen. Sections (4 μm) were fixed in methanol and blocked with 5% BSA. Anti-mouse antibodies (1:100 dilution) were added and incubated with the sections. Secondary antibodies were added after wash as indicated in the figure legends. Images were acquired using a Nikon E1000 fluorescent microscope equipped with camera (Melville, NY).
Western blot
Extraction of proteins from kidneys and purified splenic B cells for immunoblotting was performed using RIPA buffer (Santa Cruz Biotechnology, Dallas, TX). Antibodies against pStat1(Tyr701), total Stat1, pStat3(Tyr705), Stat3, Bcl-2, and β-actin were purchased from Cell Signaling Technology (Danvers, MA).
RT-PCR
Total RNA was isolated using the PureLink RNA Mini Kit (Ambion, Carlsbad, CA) from the kidney lysates. The first strand cDNA was synthesized from 1 μg of total RNA using the High Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA). Each cDNA sample was amplified by using specific FAM-NGB primers against IL-6 and IFN-γ from Life Technologies (Grand Island, NY). The housekeeping gene GAPDH was assayed in parallel, as an internal control. Real-time PCR was performed in an Applied Biosystems Real-time PCR system using TaqMan Gene Expression Master Mix. The CT of each test message was first normalized using the CT for GAPDH, assayed in the same sample. Fold change was then calculated (19).
Statistical analysis
Statistical significance was determined using Student’s t test and data are presented as means ± SD. Asterisks: *p<0.05, **p<0.01.
Results
Stat1-deficiency but not Stat2-deficiency exacerbated chronic GVH disease
IFN signaling has an important role in the development and pathogenesis of lupus. Stats have highly specific functions in the control of various immune responses. To understand the roles of Stat1 and Stat2 in the development of bm12-induced lupus like cGVH disease, we induced cGVHD in wild type (WT) C57BL/6 mice or mice lacking either Stat1 (Stat 1-KO) or Stat2 (Stat2-KO). We next measured and compared serum autoantibodies levels and proteinuria in these Stat1-, Stat2-deficient and WT recipients of bm12 alloreactive T cells. As shown in figure 1, WT and Stat2-KO mice developed similar levels of anti-dsDNA autoantibodies starting at week 2, with a decline noted after week 4 (Fig. 1A). In contrast, Stat1-KO mice showed a prolonged and significant increase in anti-dsDNA autoantibodies when compared to WT mice between week 4 and week 7 (Fig. 1B). Increased total IgG autoantibody titers were accompanied by increased proteinuria in mice lacking Stat1. Starting at week 5, proteinuria was elevated in the urine of Stat1-KO mice but not in WT mice with cGVHD (Fig. 1C). Severe autoimmune disease in Stat1-KO cGVH mice led to increased mortality (Fig. 1D), probably due to renal failure. About 40% of bm12-injected Stat1-KO mice died between week 10 and week 20, while over 90% of cGVHD WT mice survived.
Figure 1. Stat1-deficient mice develop severe chronic GVH disease.

A and B. WT, Stat1-KO mice, and Stat2-KO mice, all on C57BL/6 background, were injected i.p. with 1×108 bm12 splenocytes. Serum samples were prepared from peripheral blood of experimental mice the day of injection and at weekly intervals thereafter. Mouse sera were stored at −40°C. A-B. Anti-dsDNA titers were determined by ELISA as previously described (1, 16). C. Proteinuria levels of WT and Stat1-KO mice were determined with Uristix (Bayer Co., Elkhart, IN). D. Mouse survival was followed daily for six months. Data are representative of three experiments.
Anti-dsDNA isotype analysis revealed markedly elevated and more prolonged IgG1 and IgG2b autoantibody responses in Stat1-KO mice compared to WT mice with cGVHD (Fig. 2A and B). The diminished IgG3 response observed in Stat1-KO mice after alloreactive stimulation was probably due to the impaired IFN-γ signaling pathway (Fig. 2C), as IFN-γ has been reported to promote switching to IgG3 (20). Class switching to IgG2c was similar to the WT counterparts (Fig. 2D). Similar results were observed with anti-chromatin autoantibody isotype analysis (Data not shown). These results may reflect deregulated T cell cytokine (IL-4, IFN-γ) mediated B cell activation and antibody class switching (21). They may also indicate a role of Stat1 or Stat3 activation in response to BCR-engaged antibody production (22, 23).
Figure 2. Autoantibody isotype profiles from cGVHD Stat1-KO mice.

Chronic GVHD was induced in Stat1-KO and WT mice. Serum samples were drawn and assessed for isotype-specific anti-dsDNA autoantibodies by ELISA. Data are representative of two independent experiments.
Increased autoantibody response in Stat1-KO hosts correlates with enhanced B-cell activation
In bm12-induced cGVHD, cognate interaction of CD4 T cells with B-cell MHC II molecules results in the loss of host B cell tolerance and the production of autoantibodies typical of SLE. Host B cells show phenotypic changes indicative of diffuse activation. We, therefore, asked if increased autoantibody responses were accompanied by enhanced B-cell activation in the Stat1-KO mice. Figure 3A shows that B cells from Stat1-KO mice with cGVHD displayed an enhanced activation phenotype at weeks 4 and 6 (increased CD80, CD86, and MHC-II) compared to WT mice given the same treatment. We then purified B cells from WT and Stat1-KO mice 2 and 4 weeks after cGVHD. B-cell expression of Stat1/3 and SOCS1/3 were analyzed by Western blotting. We found about a significant increase of pStat3 (Tyr705) in B cells from Stat1-KO mice compared to B cells from WT mice (Fig. 3B). Activation of Stat3 resulted in subsequently significant expression of anti-apoptotic Bcl-2 in B cells from Stat1-KO mice compared to B cells from WT mice under the same treatment (Fig. 3B). Stat1 and Stat3 negatively regulate one another through the induction of SOCS. Stat3 and SOCS3 activation were previously reported to be enhanced in Stat1-KO mice (9). As shown in Figure 3C, SOCS1 levels in B cells were found undetectable in uninduced and cGVHD WT and cGVHD Stat1-KO mice. Levels of SOCS3 greatly increased upon the induction of cGVHD in B cells from WT and Stat1-KO mice. In contrast, WT and Stat1-KO B cells showed similar levels of SOCS3 expression when cGVHD was induced (Fig. 3C). It is important to know that B-cell expression of Stat1 and Stat3 and SOCS1 and SOCS3 was only observed at week 2 after cGVHD induction in WT and Stat1-KO mice.
Figure 3. Enhanced B-cell activation in Stat1-KO mice with cGVHD.

A. Single cell suspension of splenocytes from the cGVHD Stat1-KO and WT mice was prepared and subjected to FACS analysis. Histograms represent B cells gated on CD19 positivity. Expression of B-cells activation markers (CD80, CD86, and MHC-II) was analyzed. B-C. Purified B cells were lysed and subjected to Western blot analysis with antibodies indicated in the figure. Representative Western blots were shown. Western blots from two independent experiments were analyzed using the ImageStudio software (Li-Cor, Lincoln, NE).
Elevated immune complex deposition in the glomeruli and significantly more severe kidney damage in Stat1-KO mice with cGVHD
Because no other organ damage was observed (data not shown), it is likely that renal insufficiency caused the death of Stat1-KO mice with cGVHD. Pathological staining revealed early onset injury in the kidneys of Stat1-KO mice. Started at week 6, mild inflammation developed in the kidneys of cGVHD Stat1-KO host mice, with interstitial infiltration and hyper-cellularity (Fig. 4A). Six months after cGVHD, glomerular and tubular necrosis developed in Stat1-KO mice (Fig. 4A). One explanation is that enhanced glomerular damage in the Stat1-KO mice was due to increased deposition of nephritogenic immune complexes. IgG deposition was identified by immunofluorescent staining with anti-mouse IgG. Glomerular IgG deposition was evident in WT mice with cGVHD starting at week 2 (Fig. 4B), a trend that correlated with the autoantibody profile (Fig. 1B). Immune complex deposition was also present at week 2 in the kidney of Stat1-KO mice with cGVHD. Yet a significant and further increase in immune complex deposition in glomeruli was observed in cGVHD Stat1-KO mice starting at week 6 that was maintained until the end of study (Fig. 4B).
Figure 4. Stat1-KO mice developed more severe renal injury.
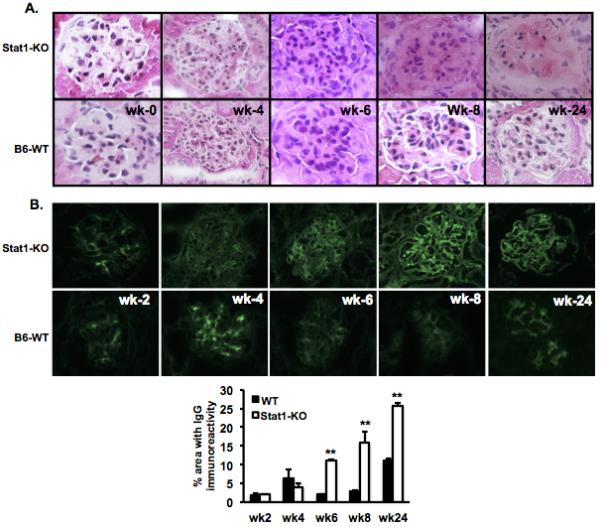
Mice were euthanized at week 2, 4, 6, 8, and 24. Kidneys were frozen in OCT with liquid nitrogen and sectioned at 4 μm. A. Sections were stained with H&E and B. FITC anti-mouse IgG. Immunostaining was quantified using a bioquantification software system (Bioquant TCW 98, Nashville, TN). The percent area fractions of the area containing thresholded pixels versus the area of the entire region of interest (ROI) are shown at the bottom and expressed as mean ±SEM (34). Data are representative of three individual experiments. Magnification: x400.
Stat3 upregulation and activation in the glomerular mesangial cells of Stat1-KO mice with cGVHD
Both type I and type II IFN can activate Stat1and data presented in Figure 1 indicate that the enhanced autoimmunity in Stat1-KO mice could be INF II pathway dependent, because the IFN-I pathway involves Stat1/Stat2 heterodimer and Stat2-KO mice developed similar autoantibody responses compared to WT mice with cGVHD. IFN-γ can activate Stat3 and its activation is exacerbated and prolonged in Stat1-null mouse embryonic fibroblasts when compared against WT cells (6). Various studies have shown Stat3 expression in glomeruli and Stat3 can be expressed and activated in the kidney by immune complexes and IL-6 (24). We therefore asked if the severe kidney inflammation observed in the cGVHD Stat1-KO mice is due to increased Stat3 expression. Compared to WT kidneys, Stat3 protein expression was in fact significantly elevated in the glomeruli of Stat1-KO mice with levels, and peaking at week 4 and week 6 (Fig. 5A and B). Anti-phospho tyrosine 705 specific antibody staining showed activation of Stat3 in the glomeruli of Stat1-KO mice with cGVHD (Fig. 5C). In contrast, Stat3 expression in the WT mice with cGVHD was barely detectable (Fig. 5A). Co-staining with glomerular cell markers revealed mesangial-specific expression of Stat3 in Stat1-KO glomeruli (Fig. 5D). Expression of Stat3 on mesangial cells was further confirmed by FACS analysis (Fig. 5E). The expression of Stat1 was detectable (week 2-8) in the WT mice with cGVHD, but to a lesser degree than Stat3 (Fig. 5A).
Figure 5. Stat3 expression in the glomerular mesangial cells of Stat1-KO mice with cGVHD.

Kidney sections (4μm) were prepared at different time points as indicated. A. Expression of Stat1 and Stat3 was revealed by staining with anti-Stat1 and anti-Stat3 antibody and visualized with a Nikon E1000 fluorescence microscope. B. Quantified levels of Stat3 immunofluorescence were shown as percent area fractions against the entire ROI as previously described. C. Stat3 phosphorylation was analyzed with anti-phosphotyrosine 705 antibody. D. Co-localization of Stat3 with mesangial cells (α-SMA), podocytes (Nephrin), and endothelial cells (CD31) was evaluated in glomeruli of Stat1-KO mice. E. FACS analyses of Stat3 on glomerular cell subsets were gated on mesangial cells. Data are representative of at least three different sections from individual mice in each experiment and were evaluated in two separate experiments. Magnification: x400.
Increased IL-6 secretion and macrophage infiltration were associated with Stat3 expression
Stat1 and Stat3 can be reciprocally activated in response to IL-6 and IFN-γ. The expression of IL-6 is increased in renal injury (25). Therefore, we stained kidney sections from WT and Stat1-KO at weeks 4 and 6 after bm12 splenocytes injection and found a significant increase of IL-6 expression in the glomeruli of Stat1-KO mice with cGVHD but not in the glomeruli of WT mice under the same conditions (Fig. 6A). Stat3 is known to be phosphorylated transiently in response to IFN-γ, but its phosphorylation level was found markedly enhanced and prolonged in Stat1-null mice (6). We hypothesized that Stat3 expression and activation observed in the kidney of Stat1-KO might be due to increased IFN-γ expression. Indeed, very strong expression of IFN-γ was found in the glomeruli of cGVHD Stat1-KO mice when compared against kidney sections from cGVHD WT mice (Fig. 6A). RT-PCR analysis also revealed increased levels of IL-6 and IFN-γ mRNA at weeks 4 and 6 after induction of cGVHD in Stat1-KO mice compared to WT mice. Moreover, the increased levels of IL-6 and IFN-γ in Stat1-KO mice kidneys were statistically significant at week 6 (Fig. 6B).
Figure 6. Increased IL-6 and IFN-γ secretion and macrophage infiltration in the glomeruli of Stat1-KO mice with cGVHD.

Kidney sections prepared from cGVHD WT and cGVHD Stat1-KO mice were incubated with A. Rabbit anti-IL6, anti-IFN-γ, and visualized with anti-rabbit Fluro488. B. Expression levels of IL-6 and IFN-γ were analyzed by RT-PCR. C. FITC-conjugated anti-CD11b antibodies were incubated with kidney sections from cGVHD WT and Stat1-KO mice. Data are representative of two independent experiments.
The IL-6/Stat3 pathway has been reported to be essential for macrophage infiltration (26), and macrophage infiltration into the kidney contributes to pathological damage. To determine whether increased IL6 and Stat3 expression in the kidney of Stat1-KO mice was associated with macrophage infiltration, we performed immunofluorescence staining on kidney sections from WT and Stat1-KO mice undergoing cGVHD. Figure 6B shows infiltration of macrophages in the kidney of Stat-KO mice compared to WT mice with cGVHD. Macrophage infiltration was limited to the blood vessel area (Fig. 6B top, right) and to the glomeruli (Fig. 6B bottom, right).
Discussion
The importance of interferons and their regulatory factors in SLE is well established (27, 28). This study of the interplay between Stat1 and Stat3 in experimental lupus further contributes to our understanding of Stats and lupus related inflammation. In particular, we showed that the absence of Stat 1 led to increased activation of Stat3 in B cells and glomerular mesangial cells in our model of lupus. Activation of Stat3 was transiently observed in B cells of WT recipient mice of allogeneic cells, while it was consistently noted that Stat1-KO hosts showed much higher degrees of B-cell activation and greater autoantibody responses. The accompanying increase in Stat3 expression was associated with acquisition of markers of diffuse B-cell activation.
These studies suggest important regulatory interactions between Stat1 and Stat3 in the control of lupus autoimmunity. Previous studies have shown reciprocal cross inhibition between Stat1 and Stat3 through SOCS1 and SOCS3 expression. Our Western blot results revealed SOCS3 expression in B cells from both WT and Stat1-KO mice with cGVHD at the same time point (week 2). However, B-cell SOCS3 levels were similar between WT and Stat1-KO mice, indicating other mechanisms may regulate Stat3-upregulation and B-cell activation. It is even possible that SOCS3 expression is a consequence of Stat3 activation, since SOCS3 is the major negative regulator of Stat3 (9).
Stat1 expression is present in renal biopsies of lupus nephritis patients and expression levels positively correlated with disease activity (29). Various studies show Stat3 activation in glomerular mesangial cells and proximal tubule cells. Stat3 can be expressed and activated in the kidney of rats with immune complex glomerulonephritis using BSA as an antigen. These rats also have increased macrophage infiltration (24). Furthermore, activating germline mutations in Stat3 cause early-onset multi-organ autoimmune disease (30). Mild kidney inflammation, which usually takes 2-3 months to develop in cGVHD WT mice, developed at week 6 in cGVHD Stat1-KO mice. Though increased autoantibody titers led to elevated immune complex deposition, it is unlikely that moderately (though statistically significant) enhanced autoantibody titers were the sole cause of the robust kidney inflammation and increased mortality rate observed in cGVHD Stat1-KO mice. Supporting this notion were studies revealing that the onset of anti-dsDNA antibody production occurred well after the onset of nephritis following pristane treatment, suggesting that these antibodies were not directly responsible for inducing renal disease (31). We hypothesize that local activation and kidney-targeted infiltration play an important role. Indeed, we observed significantly increased Stat3 expression and activation on mesangial cells. Macrophage infiltration was also evident in the kidneys of Stat1-KO mice with cGVHD.
Cytokines are known to play an important role in the pathogenesis of lupus nephritis and the Jak/Stat pathway is important in mediating signal transduction of cytokines. Stat3 activation can be induced by multiple cytokines, such as IL-6 and IFN-γ. IFN-γ is a potent activator of Stat1, while IL-6 primarily activates Stat3 (32). However, IFN-γ-induced Stat3 phosphorylation is enhanced and prolonged in Stat1-KO mice (6). Our finding of Stat3 expression/activation in the kidney strongly supports this notion. Stat3 has been implicated in the inflammatory proliferation of cells. In our cGVHD model, expression of Stat3 in the mesangial cells of the Stat1-KO mice was associated with mesangial hyper-cellularity. Inhibiting renal expression of IFN-γ is accompanied by reduced infiltration of macrophages and attenuated deposition of IgG in the kidneys (33). Increased levels of IFN-γ in the glomeruli of Stat1-KO mice were associated with macrophage infiltration when cGVHD was induced. Although IgG deposition was associated with globally enhanced autoantibody responses in the cGVHD Stat1-KO mice, the increased macrophage infiltration may be attributed to increased IFN-γ expression in the kidney of cGVHD Stat1-KO mice. Macrophage (CD11b+) infiltration may cause further damage to the already inflamed kidney of cGVHD Stat1-KO mice.
Presently, various inhibitory strategies to block Stat signaling and function are being pursued. MRL/lpr mice treated with the selective Jak2 inhibitor (AG490, which inhibits both Stat1 and Stat3) had significantly reduced proteinuria and improved renal function (33). In this regard, inhibition of both Stat1 and Stat3 signaling may hold therapeutic potential for LN. Our studies emphasize that there are complex interactions between different Stats that need to be considered as Jak-directed therapy becomes broadly accepted, because of the wide variety of cells that express Stats and the interest in exploiting them becomes more and more tantalizing.
Acknowledgement
We are grateful to Dr. Fred D. Finkelman for the critical reading of the manuscript and to Dr. Mark Birkenbach for assistance with the evaluation of kidney histology and pathology.
This work was supported by NIH grants (R21AR065692 to P.L.C; DK095067 to W.H.S.) and a grant from the Alliance for Lupus Research (P.L.C.).
Abbreviations
- SLE
systemic lupus erythematosus
- cGVHD
chronic graft versus host disease
- Stats
signal transducer and activator of transcription
- IFN-I
type I interferon
- SOCS
suppressor of cytokine signaling
References
- 1.Tsokos GC. Systemic lupus erythematosus. The New England journal of medicine. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 2.Cozzani E, Drosera M, Gasparini G, Parodi A. Serology of Lupus Erythematosus: Correlation between Immunopathological Features and Clinical Aspects. Autoimmune diseases. 2014;2014:321359. doi: 10.1155/2014/321359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koutsokeras T, Healy T. Systemic lupus erythematosus and lupus nephritis. Nature reviews. Drug discovery. 2014;13:173–174. doi: 10.1038/nrd4227. [DOI] [PubMed] [Google Scholar]
- 4.Eisenberg RA, Via CS. T cells, murine chronic graft-versus-host disease and autoimmunity. Journal of autoimmunity. 2012;39:240–247. doi: 10.1016/j.jaut.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nature reviews. Immunology. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 6.Qing Y, Stark GR. Alternative activation of STAT1 and STAT3 in response to interferon-gamma. The Journal of biological chemistry. 2004;279:41679–41685. doi: 10.1074/jbc.M406413200. [DOI] [PubMed] [Google Scholar]
- 7.Nowling TK, Gilkeson GS. Mechanisms of tissue injury in lupus nephritis. Arthritis research & therapy. 2011;13:250. doi: 10.1186/ar3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arakawa T, Masaki T, Hirai T, Doi S, Kuratsune M, Arihiro K, Kohno N, Yorioka N. Activation of signal transducer and activator of transcription 3 correlates with cell proliferation and renal injury in human glomerulonephritis. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2008;23:3418–3426. doi: 10.1093/ndt/gfn314. [DOI] [PubMed] [Google Scholar]
- 9.Hong F, Jaruga B, Kim WH, Radaeva S, El-Assal ON, Tian Z, Nguyen VA, Gao B. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. The Journal of clinical investigation. 2002;110:1503–1513. doi: 10.1172/JCI15841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity. 2009;31:539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stephanou A, Latchman DS. Opposing actions of STAT-1 and STAT-3. Growth factors. 2005;23:177–182. doi: 10.1080/08977190500178745. [DOI] [PubMed] [Google Scholar]
- 12.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nature reviews. Molecular cell biology. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 13.Levy DE, Lee CK. What does Stat3 do? The Journal of clinical investigation. 2002;109:1143–1148. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, Lyons JJ, Engelhardt KR, Zhang Y, Topcagic N, Roberson ED, Matthews H, Verbsky JW, Dasu T, Vargas-Hernandez A, Varghese N, McClain KL, Karam LB, Nahmod K, Makedonas G, Mace EM, Sorte HS, Perminow G, Rao VK, O'Connell MP, Price S, Su HC, Butrick M, McElwee J, Hughes JD, Willet J, Swan D, Xu Y, Santibanez-Koref M, Slowik V, Dinwiddie DL, Ciaccio CE, Saunders CJ, Septer S, Kingsmore SF, White AJ, Cant AJ, Hambleton S, Cooper MA. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. 2015;125:591–599. doi: 10.1182/blood-2014-09-602763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisenberg R. The chronic graft-versus-host model of systemic autoimmunity. Current directions in autoimmunity. 2003;6:228–244. doi: 10.1159/000066864. [DOI] [PubMed] [Google Scholar]
- 16.Shao WH, Eisenberg RA, Cohen PL. The Mer receptor tyrosine kinase is required for the loss of B cell tolerance in the chronic graft-versus-host disease model of systemic lupus erythematosus. Journal of immunology. 2008;180:7728–7735. doi: 10.4049/jimmunol.180.11.7728. [DOI] [PubMed] [Google Scholar]
- 17.Vielhauer V, Anders HJ, Perez de Lema G, Luckow B, Schlondorff D, Mack M. Phenotyping renal leukocyte subsets by four-color flow cytometry: characterization of chemokine receptor expression. Nephron Exp Nephrol 93: e63. 2003 doi: 10.1159/000068517. [DOI] [PubMed] [Google Scholar]
- 18.Shao WH, Kuan AP, Wang C, Abraham V, Waldman MA, Vogelgesang A, Wittenburg G, Choudhury A, Tsao PY, Miwa T, Eisenberg RA, Cohen PL. Disrupted Mer receptor tyrosine kinase expression leads to enhanced MZ B-cell responses. Journal of autoimmunity. 2010;35:368–374. doi: 10.1016/j.jaut.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao WH, Zhen Y, Rosenbaum J, Eisenberg RA, McGaha TL, Birkenbach M, Cohen PL. A protective role of Mer receptor tyrosine kinase in nephrotoxic serum-induced nephritis. Clinical immunology. 2010;136:236–244. doi: 10.1016/j.clim.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snapper CM, McIntyre TM, Mandler R, Pecanha LM, Finkelman FD, Lees A, Mond JJ. Induction of IgG3 secretion by interferon gamma: a model for T cell-independent class switching in response to T cell-independent type 2 antigens. J Exp Med. 1992;175:1367–1371. doi: 10.1084/jem.175.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bossie A, Vitetta ES. IFN-gamma enhances secretion of IgG2a from IgG2a-committed LPS-stimulated murine B cells: implications for the role of IFN-gamma in class switching. Cell Immunol. 1991;135:95–104. doi: 10.1016/0008-8749(91)90257-c. [DOI] [PubMed] [Google Scholar]
- 22.Karras JG, Huo L, Wang Z, Frank DA, Zimmet JM, Rothstein TL. Delayed tyrosine phosphorylation and nuclear expression of STAT1 following antigen receptor stimulation of B lymphocytes. Journal of immunology. 1996;157:2299–2309. [PubMed] [Google Scholar]
- 23.Wang L, Kurosaki T, Corey SJ. Engagement of the B-cell antigen receptor activates STAT through Lyn in a Jak-independent pathway. Oncogene. 2007;26:2851–2859. doi: 10.1038/sj.onc.1210092. [DOI] [PubMed] [Google Scholar]
- 24.Zhang W, Chen X, Shi S, Wei R, Wang J, Yamanaka N, Hong Q. Expression and activation of STAT3 in chronic proliferative immune complex glomerulonephritis and the effect of fosinopril. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2005;20:892–901. doi: 10.1093/ndt/gfh652. [DOI] [PubMed] [Google Scholar]
- 25.Lemay S, Rabb H, Postler G, Singh AK. Prominent and sustained up-regulation of gp130-signaling cytokines and the chemokine MIP-2 in murine renal ischemia-reperfusion injury. Transplantation. 2000;69:959–963. doi: 10.1097/00007890-200003150-00049. [DOI] [PubMed] [Google Scholar]
- 26.Zhang C, Li Y, Wu Y, Wang L, Wang X, Du J. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. The Journal of biological chemistry. 2013;288:1489–1499. doi: 10.1074/jbc.M112.419788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z, Davidson A. IFNalpha Inducible Models of Murine SLE. Front Immunol. 2013;4:306. doi: 10.3389/fimmu.2013.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obermoser G, Pascual V. The interferon-alpha signature of systemic lupus erythematosus. Lupus. 2010;19:1012–1019. doi: 10.1177/0961203310371161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Lostao L, Ordi-Ros J, Balada E, Segarra-Medrano A, Majo-Masferrer J, Labrador-Horrillo M, Vilardell-Tarres M. Activation of the signal transducer and activator of transcription-1 in diffuse proliferative lupus nephritis. Lupus. 2007;16:483–488. doi: 10.1177/0961203307079618. [DOI] [PubMed] [Google Scholar]
- 30.Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Lango Allen H, De Franco E, McDonald TJ, Rajala H, Ramelius A, Barton J, Heiskanen K, Heiskanen-Kosma T, Kajosaari M, Murphy NP, Milenkovic T, Seppanen M, Lernmark A, Mustjoki S, Otonkoski T, Kere J, Morgan NG, Ellard S, Hattersley AT. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet. 2014;46:812–814. doi: 10.1038/ng.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richards HB, Satoh M, Shaw M, Libert C, Poli V, Reeves WH. Interleukin 6 dependence of anti-DNA antibody production: evidence for two pathways of autoantibody formation in pristane-induced lupus. J Exp Med. 1998;188:985–990. doi: 10.1084/jem.188.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stephanou A, Latchman DS. STAT-1: a novel regulator of apoptosis. Int J Exp Pathol. 2003;84:239–244. doi: 10.1111/j.0959-9673.2003.00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S, Yang N, Zhang L, Huang B, Tan H, Liang Y, Li Y, Yu X. Jak/STAT signaling is involved in the inflammatory infiltration of the kidneys in MRL/lpr mice. Lupus. 2010;19:1171–1180. doi: 10.1177/0961203310367660. [DOI] [PubMed] [Google Scholar]
- 34.Al-Shatti T, Barr AE, Safadi FF, Amin M, Barbe MF. Increase in inflammatory cytokines in median nerves in a rat model of repetitive motion injury. J Neuroimmunol. 2005;167:13–22. doi: 10.1016/j.jneuroim.2005.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]