

Abstract
Esophageal squamous cell carcinoma (ESCC) has a high mortality rate. To determine the molecular basis of ESCC development, this study sought to identify characteristic genome-wide alterations in ESCC, including exonic mutations and structural alterations. The clinical implications of these genetic alterations were also analyzed. Exome sequencing and verification were performed for nine pairs of ESCC and the matched blood samples, followed by validation with additional samples using Sanger sequencing. Whole-genome SNP arrays were employed to detect copy number alteration (CNA) and loss of heterozygosity (LOH) in 55 cases, including the nine ESCC samples subjected to exome sequencing. A total of 108 non-synonymous somatic mutations (NSSMs) in 102 genes were verified in nine patients. The chromatin modification process was found to be enriched in our gene ontology (GO) analysis. Tumor genomes with TP53 mutations were significantly more unstable than those without TP53 mutations. In terms of the landscape of genomic alterations, deletion of 9p21.3 covering CDKN2A/2B (30.9%), amplification of 11q13.3 covering CCND1 (30.9%), and TP53 point mutation (50.9%) occurred in two-thirds of the cases. These results suggest that the deregulation of the G1 phase during the cell cycle is a key event in ESCC. Furthermore, six minimal common regions were found to be significantly altered in ESCC samples and three of them, 9p21.3, 7p11.2, and 3p12.1, were associated with lymph node metastasis. With the high correlation of TP53 mutation and genomic instability in ESCC, the amplification of CCND1, the deletion of CDKN2A/2B, and the somatic mutation of TP53 appear to play pivotal roles via G1 deregulation and therefore helps to classify this cancer into different genomic subtypes. These findings provide clinical significance that could be useful in future molecular diagnoses and therapeutic targeting.
Keywords: Esophageal squamous cell carcinoma, Genomic subtype, Somatic mutation, Copy number alteration, Cell cycle deregulation
Introduction
Esophageal cancer is a common type of cancer that is strongly associated with high mortality and ranks as the sixth leading cause of death from cancer [1]. Squamous cell carcinoma and adenocarcinoma are the major types of esophageal cancer. In the area with high prevalence of esophageal cancer (also known as the “esophageal cancer belt”), which stretches from Northern Iran through the Central Asia to the North Central China, 90% of all cases are diagnosed as squamous cell carcinomas [2–4]. The 5-year survival rate for esophageal squamous cell carcinoma (ESCC) has been low, but the principal causes for this disease remain elusive [5–7]. Previous studies have demonstrated a series of genetic alterations, including a high somatic mutation rate for TP53 and genomic instability in numerous chromosomes [8–13]. A comprehensive description of various types of genetic alterations in ESCC and their correlation with clinical outcome would be a great step forward in our understanding of the mechanism involved in ESCC development, and could be applied to improve the survival rate of patients.
In this study, we analyzed the ESCC genome by conducting exome sequencing of nine ESCC sample pairs along with whole-genome SNP arrays of 55 tumor samples in total. Our results revealed a very high correlation of TP53 somatic mutation with genomic instabilities in ESCC. Interruption of G1 control by TP53 somatic mutation and copy number alterations (CNAs) was found in over 65% of ESCC cases. Furthermore, for the first time we have identified a significant correlation between copy number aberrations in three minimal common regions (MCRs), i.e., amplification of 7p11.2, deletion of 3p12.1, and deletion of 9p21.3, and lymph node metastases in ESCC (P < 0.05).
Results
High heterogeneity of the somatic mutation spectrum in ESCC
To identify the mutation profile in ESCC, exome sequencing was conducted for tumor and the matched blood samples from nine ESCC patients (further information about the patients enrolled in this study is described in Materials and methods, Figures S1 and S2, and Table S1). As summarized in Table S2, after quality control, we obtained 31 × coverage on average for the targeted bases and over 93% of all bases were covered in the targeted regions. The non-synonymous somatic mutations (NSSMs, present in tumor but absent in blood samples) identified by exome sequencing were validated using either Sequenom MassARRAY or Sanger sequencing in both tumor and blood samples. Overall, 108 NSSMs in 102 genes were verified (Table S3). The number of somatic mutations per tumor sample was highly variable (range 0–36 per sample, no somatic mutation identified in 3 samples). The most common type of mutations was missense (80, 74.07%), as compared with other types of mutations such as nonsense mutations (12, 11.11%) and small indels (14, 12.96%). Only one single splice-site (0.93%) and one read-through mutations (0.93%) were detected, respectively.
To screen for recurrently-mutated genes, we looked into the genes with mutations in two or more of the nine tumor samples (Figure S3). As a result, somatic mutations in TP53 were identified in five tumor samples (5/9, 55.6%). Both FBXL4 and DMD were mutated in two tumor samples. DMD was excluded from our further analysis since this is the largest gene (measuring 2.4 Mb in human genome according to RefSeq summary). In general, random mutations may occur more frequently in larger genes, as supported by our observation of numerous mutations detected in DMD in non-cancerous tissues and in other datasets. TP53 and FBXL4 were subjected to Sanger sequencing in additional 46 and 120 samples, respectively (TP53 and Panel 2, Figure S2). 50% of the validated samples (23/46) were found to carry at least one NSSM in TP53. Totally 26 somatic mutations were identified in these 23 patients, among which 3 patients carried two different mutations in TP53. These included 19 missense mutations, 5 truncations, and 2 single nucleotide insertions that possibly result in frameshift. The possible consequences of the 19 missense mutations were predicted using PolyPhen 2 [14] and are listed in Table S4. Notably, at least one TP53 mutation in each of the 23 patients was predicted to be deleterious. Only one of the 26 somatic mutations in TP53 was reported in dbSNP, a database of single nucleotide polymorphisms, with a very low frequency (rs201382018, identified in patient 109596, the allele frequency is 0.02% or 1/5008, accordingly). With inclusion of the discovery set, somatic mutations in TP53 were observed in 28/55 of ESCCs (50.90%, Tables S3 and S4). Despite the fact that there was no particular hot spot identified, most somatic mutations were localized in exons 4–8 of TP53. On the other hand, only two somatic missense mutations were found in FBXL4 in the additional 120 samples of ESCCs, resulting in a total mutation rate of 3.1% in all the samples examined.
Furthermore, we conducted Sanger sequencing on the coding regions of several genes for the validation samples. These genes were selected based on their known cellular functions or their roles in various cancers. First, we examined FBXW7, CD40LG, ANG, and INHBC (Panel 2), WNT2B (Panel 3), and XRCC2 (Panel 5) as shown in Figure S2, because mutations in these genes were detected in the discovery sample set (one out of nine patients, Table S3). However, no mutation was seen in the additional 120 samples. Next, we examined mutational regions or “hot spots,” including exon 4 of AKT1, exons 15 and 19 of BRAF, and exons 9 and 20 of PIK3CA in 120 cases (Panel 4, Figure S2). We failed to identify any somatic mutations in the regions examined either.
Taken together, the somatic mutation spectrum showed very high heterogeneity in ESCC between different tumor samples. With the exception of TP53, some known cancer-related genes, including FBXW7, AKT1, BRAF, and PIK3CA, showed very low mutation rates in our sample population (up to 120 samples).
Mutations involved in chromatin modification process
To explore the biological processes that were interrupted in ESCC patients, we performed a GO cluster analysis on all the somatically-mutated genes detected in this study. Ten genes involved in chromosome organization were significantly clustered (P = 0.002, Table S5). In particular, seven of them (identified in five patients), including CHD3, MLL, NASP, PHF16, SMARCD3, TSPYL2, and UBN1, were further enriched in the chromatin modification subset (P = 0.005). Among these seven missense mutations, six mutations were predicted by SIFT and PolyPhen 2 to have a deleterious effect on the protein function (Table S3), suggesting that there is a frequent disruption of the chromatin modification processes in the pathogenesis of ESCC.
Considering that mutations of chromatin remodeling genes have been highlighted in several cancer studies [15–19], we then conducted Sanger sequencing for all the coding regions of these seven genes in different validation sets (samples for validation were selected randomly, however, due to limited volume of DNA samples of each patient, we cannot test all genes in a single validation set). CHD3, MLL, NASP, PHF16, and UBN1 were tested in 16 samples (Panel 1), whereas TSPYL2 and SMARCD3 were tested in 120 samples (Panel 3) as indicated in Figure S2. However, no additional mutations in any of the validation samples were detected. This is most likely due to insufficient number of genes tested, in view of the fact that there are over 270 genes involved in chromatin modification, according to GO annotation.
A complex landscape of structural alterations
We used allelic imbalance as an indicator of chromosomal alterations in ESCC. A total of 107 regions of allelic imbalance were identified using window sliding, with estimated sizes in the range of 2–241 Mb (Figure S4). Two major patterns were found in these nine ESCCs, as illustrated in Figure 1. Tumor samples from patients 99648, 100036, and 102995 appeared to suffer few or no chromosomal alterations. On the other hand, the remaining 6 tumor samples showed many regions of allelic imbalance, including a few large regions. For instance, in the tumor sample from patient 101919, 22 allelic imbalance regions were identified, and the region located at Chromosome 8 measured ∼144 Mb, almost covering the whole chromosome.
Figure 1.

Allelic imbalance in nine ESCC sample pairs detected using exome sequencing
Bar plots illustrating the minor allele fraction (MAF) of informative SNPs in each exome of blood and tumor samples. The X-axis indicates the genomic position of each informative SNP on autosomes. The imbalanced regions revealed by window-sliding method are indicated in red. Apparently tumor samples from patients 99648, 100036, and 102995 were rarely affected by any alteration characterized by allelic imbalance as seen in the other six tumor samples.
Considering the difficulty in determining the boundary and copy number status in exome sequencing data, we next performed SNP arrays on 55 tumors and 9 blood samples, including the 9 tumors and 7 blood samples that were evaluated by exome sequencing, in order to further characterize the genomic aberrations in ESCC. The fraction of copy number gain, copy number loss, and copy number neutral loss of heterozygosity (CNNLOH) were then calculated in order to estimate the genome instability in each sample.
As shown in Table S6, the fraction of genomic instability, including copy number gain, copy number loss, and CNNLOH, ranged from 0.001 to 0.97 (median, 0.557) among the 55 tumor samples. In total, eight tumor samples showed a genomic fraction of structural variation of less than 0.05, including the three tumor samples that showed very low allelic imbalance in exome sequencing as shown in Figure 1. Moreover, 30 of the 55 tumors (54.5%) had genome-wide alteration fractions higher than 0.5 (Table S6), suggesting severe genomic instability in those ESCC samples. In contrast to the tumors, the median fraction of altered regions in the blood samples tested was 1.8 × 10−4 (in the range of 0–0.06, Table S7), suggesting very minor, if any, genomic alterations in germ lines at the scale in this study. This was also demonstrated by an analysis using cnvPartition (a plug-in for copy number variation analysis of Genome Studio) (Figure S5 and Table S8).
Regarding the type of genomic alterations in ESCC, the median fraction of CNNLOH across the entire genome in all 55 samples was 0.29 (in the range 0–0.67), suggesting frequent occurrences of acquired uniparental disomy during mitosis. The median overall genome-wide fractions for copy number gain and loss were 0.09 (in the range of 0–0.36) and 0.05 (in the range of 6 × 10−5–0.38), respectively (Table S6). Among the 55 tumor samples examined, chromosome 3q harbored the largest fraction of copy number gain, whereas chromosome 9p had the largest fraction of copy number loss and chromosome 17p showed the largest fraction of CNNLOH (Table S9 and Figure S6).
Both TP53 mutations and CNAs point to cell cycle deregulation
Among the 55 tumor samples analyzed, we identified four significantly-amplified MCRs containing 34 known genes, as well as two deleted MCRs covering 19 genes (Figure 2, Table S10). Among them, MCRs on 11q13.3 and 9p21.3 were the most significantly amplified and deleted regions, respectively (Q = 2.41 × 10−7 and 2.76 × 10−8; 17/55 and 17/55, respectively). This resulted in the amplification of several oncogenes, including CCND1, FGF3, FGF4, and FGF19, and the deletion of the tumor suppressor genes CDKN2A and CDKN2B. In addition, the region with focal amplification in 11q22.1 (Q = 1.25 × 10−6, 9/55) contains several cancer-related genes, including YAP1, which has been reported in the liver and colorectal cancers [20–22], as well as BIRC2 and BIRC3, which activate the NF-κB signaling pathway (KEGG pathway database, http://www.genome.jp/kegg/pathway.html). Additionally, EGFR was amplified in the MCR of 7p11.2, whereas CADM2, which has been identified as a tumor suppressor gene in prostate cancer [23,24], was deleted in the MCR of 3p12.1.
Figure 2.

Profiling of genomic deletions and amplifications in 55 ESCC samples
The log R ratios of SNPs were segmented by R‐GADA algorithm [60]. The peaks in red and blue represent the GISTIC Q value (bottom) and G-score (top) of amplified regions and deleted regions, respectively. The green vertical lines indicate the Q value considered as significant in the analysis (0.05). Across autosomes 1–22, six MCRs, including 2 deletions and 4 amplifications, were identified (Q < 0.05). Genes that have been reported to be related to either ESCC or other cancers in previous studies are listed in each MCR. The MCR in the HLA region of chromosome 6 (asterisk) was not included in this study due to its extremely high degree of polymorphism. Chromosomes are represented in white (odd-numbered chromosomes) and gray (even-numbered chromosomes) rectangles alternately (with heights in proportion to the lengths of the respective chromosomes). Positions for centromeres are indicated by the dotted lines. R, probe intensity in the SNP array; MCR, minimal common region; HLA, human leukocyte antigen.
Taken together, we saw a clear grouping in ESCC cases that were enrolled in this study (Figure 3). About 2/3 of the samples carried TP53 somatic mutations or focal CNAs that make a large contribution to damaging cell cycle regulation. As demonstrated in Figure 4, the most affected nodes are at p16INK4A/p15INK4B, cyclin D1, and p53, which were caused by focal deletion of CDKN2A/2B (30.9%), focal amplification of CCND1 (30.9%), and point mutations in TP53 (50.9%), respectively. Among the 55 cases examined, 65.45% (36/55) of the patients carried at least one of these alterations. Considering the functions of p16INK4A and cyclin D1 in G1 progression, as well as the role that p53 plays in mitotic check points, cell cycle regulation appears to be greatly disrupted in tumor cells of ESCC.
Figure 3.

Occurrence of TP53 mutations and CNAs of six MCRs in 55 samples
Each solid bar in the same column represents a single case with color codes as green for point mutation, red for amplification, blue for deletion, and light gray for no alteration as indicated. The red box indicates 65% (36/55) of samples containing either the TP53 nonsynonymous mutations (in 28 samples), the amplification of 11q13.3 containing CCND1 (in 17 samples), or the deletion of 9p21.3 containing CDKN2A/CDKN2B (in 17 samples). CNA, copy number alteration.
Figure 4.

Genomic alterations point to cell cycle G1 deregulation
Both TP53 mutations and CNAs point to G1 deregulation in around two-thirds of ESCC tumor samples. Numbers of samples with TP53 somatic mutation, focal amplification, and focal deletion are labeled in green, red, and blue, respectively.
ESCCs with TP53 mutations are more genomically unstable
We found a high correlation of TP53 mutations with genome instability. As depicted in Figure 5, the median fractions of copy number gain, copy number loss, and CNNLOH in samples with TP53 mutations were 0.13, 0.11, and 0.35, respectively. These values were significantly higher than those seen in samples with wild-type TP53, which were 0.05, 0.03, and 0.08, respectively (Mann–Whitney U test P = 1.18 × 10−3, 1.43 × 10−3, and 9.65 × 10−4, respectively). In particular, 75% of the patients with TP53 mutations had severe genomic instability, while only 33% of the patients with wild-type TP53 had severe genomic instability (P = 0.002).
Figure 5.

Association of TP53 somatic mutations and genomic instability
Violin plots show the genomic alterations including copy number gain (A, P = 1.18 × 10−3), copy number loss (B, P = 1.43 × 10−3), and CNNLOH (C, P = 9.65 × 10−4) in ESCC tumors with (TP53 MUT) and without (TP53 WT) TP53 somatic mutations. The overall fraction of genomic alterations is shown in panel D (P = 1.10 × 10−4). CNNLOH, copy number neutral loss of heterozygosity. Mann–Whitney U test was performed to determine the significant differences.
Chromothripsis likely occurs in one tumor sample
Shattering of chromosomes or chromosome arms was reported in a fraction of cancer samples, which is believed to be the driver event in these cases [25]. In the SNP array screen of 55 tumor samples, 42 break points were observed on the chromosome arm 3q in one tumor sample (patient ID 111820; 1/55 or 1.8%), as demonstrated in Figure 6. This suggests the occurrence of a genomic instability-generating phenomenon known as chromothripsis, where tens to hundreds of chromosomal rearrangements occur in a “one-off” cellular event [25]. Currently, the clinical consequences of this low-prevalence genomic event in ESCC remain unknown due to the limited sample size in our study.
Figure 6.

Occurrence of chromothripsis in one ESCC tumor sample
The genomic evidence for chromothripsis in one ESCC case (patient ID 111820) is shown by comparison of tumor sample (A) with the matched blood sample (B). Both B allele frequency and log R ratio on the chromosome arm 3q of the tumor sample demonstrated numerous break points with limited copy number alterations.
Correlation of genomic alterations with clinical outcome
We also analyzed the association of genomic instability with clinical information (gender, age, cancer stage and differentiation, tobacco and alcohol consumption, and family cancer history) and survival status. Patients with severe genomic instability (⩾50% of the genome) had a higher percentage of lymph node metastasis than patients with a low (<50% of the genome) genomic instability (63.3% vs. 36.7%, P = 0.021, Table S11). In addition, patients with a higher degree of overall genomic instability showed poorer survival, although the difference was not statistically significant (P = 0.083, Figure S7). Moreover, among the six MCRs identified, three regions were significantly correlated with lymph node metastasis, including the amplification of 7p11.2 (P = 0.042), deletion of 3p12.1 (P = 0.030), and deletion of 9p21.3 (P = 0.033) (Figure 2; Table S12). On the other hand, although non-synonymous TP53 mutations occurred more frequently, we observed no correlation between these mutations and clinical outcome (data not shown).
Discussion
In this study, we performed exome sequencing on nine pairs of ESCC samples, followed by an analysis of structural alterations using SNP arrays. Due to the relatively low coverage of exome sequencing, we eliminated all of the false positive calls through Sequenom or Sanger sequencing verification. Although we could not avoid certain false negative calls, the very similar mutation rates of TP53 in the discovery set (5/9) and validation set (23/46) indicate that our exome sequencing data are reliable. Moreover, we re-sequenced five exons from three frequently-mutated cancer genes, AKT1, PIK3CA, and BRAF, in an additional 120 ESCC samples. The inability to detect somatic mutations also suggests that high frequency mutations are seldom seen in ESCC other than in TP53. This is consistent with the findings of recent genomic studies of ESCC, in which TP53 was found to be the most frequently mutated gene (>60% in all studies) [26–29]. Other potential driver genes defined by these studies mutated only in ∼20% samples at most, which were identified as singleton in our exome sequencing study, for instance somatic frameshift indel identified in ADAM29 from patient 101919.
Exome sequencing showed limited resolution of structural alterations, as indicated by the allelic imbalance (Figure 1). Therefore, to characterize these alterations more precisely, we conducted a whole-genome SNP array using both the discovery sample set and an additional set of 46 tumors. To ensure detection of structural alterations in cancer cells, we set our criterion for a segment containing at least 50 continuous SNPs. In this way, we neglected smaller germline copy number variations. Using a scanning scale of roughly 150 kb (one SNP per 3 kb on average), we observed striking evidence of genomic instability (including copy number gain, loss, and neutral LOH) than that in the blood samples. As a result, our SNP array data support that the allelic imbalance in our exome sequencing data reveals valuable information regarding structural alterations.
Deregulation of cell cycle, especially in G1 phase, plays an important role in ESCC
A number of in vitro and in vivo experiments have demonstrated the key role of cell cycle deregulation in tumorigenesis [30–33]. Many cell cycle-related factors function as either tumor suppressors or oncogenes [34–36]. The most important molecule in this process is TP53, whose high mutation rate has been reported in various types of cancer (IARC TP53 Database, http://p53.iarc.fr/). In this study, we showed strong new evidence for the old story of cell cycle deregulation in ESCC. Somatic mutations in TP53 plus two MCRs (deletion of CDKN2A/2B and amplification of CCND1) point primarily to cell cycle deregulation in over 65% of the cases examined. Therefore, TP53 mutations, which disrupt cell cycle check points at the G1/S and G2/M transitions, and the amplification of an oncogene (CCND1) or the deletion of suppressor genes (CDKN2A/2B) strongly suggest that the disruption of G1 control is a key event in the development of ESCC (Figure 4). Although the high rate of TP53 mutation and the complex DNA alterations have been observed in ESCC and other cancers [37–41], to our knowledge this is the first analysis on a large sample size that pinpoints the high frequency of G1 control deregulation caused by genomic alterations in ESCC.
TP53 somatic mutations are correlated with severe genomic instability
We observed a very high correlation between somatic mutations in TP53 and genomic instability, which has been observed in many previous studies [42–45]. In tumors with a genomic instability fraction <0.10, only 1 out of 14 samples had a mutated TP53. Conversely, in samples with fraction >0.90 for structural alteration, six out of seven samples had TP53 mutations. One assumption is that the TP53 mutations and genomic alteration may occur independently and that TP53 mutations result in a failure to undergo apoptosis in cells carrying genomic abnormalities [46–48]. On the other hand, TP53 has also been proposed to be the “genome guardian” and mutations in this gene may therefore directly cause other genomic alterations [33,49]. In our ESCC cases, despite the high mutation rate of TP53, we found no significant association between TP53 mutation and tumor stage, whereas genomic instability correlated with lymph node metastasis. Therefore, we speculate that the TP53 mutation may occur early during ESCC development, and one of the consequences is genome instability, as suggested by many previous studies [50,51].
Diverse genomic patterns reveal other potential drivers in ESCC development
In this study, we found 19 samples without any alteration in cell cycle regulation. Based on their genome instability status, these cases can further be classified into three types. Two of the 19 samples had severe genomic instability (>0.5) but contained none of the six MCRs that were identified in other individuals. Both samples shared focal amplifications at 8p11.21, which encompasses IKBKB, a node in the NF-κB pathway. Another nine samples had medium genome-wide instability with alteration fractions of 0.05–0.35. However, we failed to identify any major common patterns of somatic mutations or structural alterations in these nine tumors. More interestingly, the remaining 8 samples showed extremely low fractions of genomic instabilities (<0.05). Three out of these 8 samples were analyzed by exome sequencing with no somatic mutations detected. Although we cannot rule out the possibility of a few false negatives, these examples suggest a special category of ESCC that carries low numbers of structural variations. It is therefore, of particular interest to further study ESCC that has a relatively stable genome. Furthermore, these findings may provide fundamental insights into the molecular classification of ESCC.
Other cancer-related mutations and pathway alterations occur in ESCC
In addition to cell cycle-related copy number gains, several other genes were also amplified by the 11q13.3 focal change, such as FGF3/4/19 and CTTN. FGF3/4/19 gene cluster encodes fibroblast growth factor 3, 4, and 19, members of the FGF family. Considering their activities in the MAPK and PI3K-Akt signaling pathways, these amplified factors may also be involved in mitogenic and cell survival in ESCC. A few over-expressed genes in cancer are also located in the amplified region of 11q13.3, including MYEOV (myeloma over expressed) [52], ORAOV1 (oral cancer over expressed gene 1) [53], ANO1 in head and neck squamous cell carcinoma [54], and CTTN in breast cancer and squamous cell carcinomas of the head and neck [55,56]. Therefore, although CCND1 was considered as the major player in this MCR, the amplification of other genes may also contribute to ESCC development.
Furthermore, despite the fact that no common point mutations other than TP53 were found in our study, we observed one recurrently-mutated gene, FBXL4, in both the discovery (2/9) and validation sets (2/120). The mutation rate was 3.1% for this gene encoding F-box protein, which is one subunit of the ubiquitin protein ligase complex, implying that there is disruption in the ubiquitination in some ESCC samples. These observations call for further investigation of the ubiquitin-related protein degradation pathway in ESCC patients.
Clinical significance of our findings
In this study, we observed an association of overall tumor genomic instability and clinical outcome. In particular, we discovered that three MCRs, amplification of 7p11.2, deletion of 3p12.1, and deletion of 9p21.3, correlated with lymph node metastasis. Several known cancer-related genes, including EGFR and CDKN2A/2B, are harbored in these regions. Additionally, the function of a few other genes, such as GBE1 and CADM2 in 3p12.1, has not been intensively studied in cancers and thus deserve further investigation. These findings suggest that carriers of these MCRs may have poor disease progression and therefore, detection of these alterations may be helpful in future disease monitoring.
Furthermore, as the heterogeneity has been widely accepted as a common feature in cancer, a clinic challenge is the therapeutic strategy regarding how to deliver proper and individualized treatments. We observed a clear classification in ESCC cases as the G1 deregulation occurred in 2/3 of the cases examined. Such a high deregulation frequency in certain pathways provides promising targeting strategies for future individualized diagnoses and therapeutic development. In particular, the CDK inhibitors that target early G1 phases may be beneficial for this group of patients.
Conclusions
In this study, we found a ∼50% prevalence of TP53 mutations in ESCC, and the somatic mutation of TP53 is highly correlated with genomic instability. Three MCRs are associated with lymph node metastasis. The amplification of CCND1 and the deletion of CDKN2A/2B, together with TP53 mutations, may play pivotal roles in ESCC by deregulating G1 cell cycle signaling, which classify this cancer into different groups.
Materials and methods
Sample collection
All ESCC patients enrolled in this study were diagnosed at the Anyang Cancer Hospital (Henan Province, China) in 2007–2009. Tumor specimens and paired blood samples were collected from ESCC patients. Written informed consent was obtained before sample collection. Tumor specimens from ESCC cases were stored at −70 °C immediately following collection. Genomic DNA was purified from tumor samples using a Biomek 3000 automated workstation with a E.Z.N.A Mag-Bind Tissue DNA Kit (Omega Bio-Tek, Norcross, GA, USA), whereas DNA was extracted from blood samples using a Whole Blood DNA Extraction Kit (BioTeke, Beijing, China). DNA quality and quantity were determined using a NanoDrop 1000 (Thermo Scientific, Wilmington, DE, USA).
Demographic data and patient information, including age, gender, alcohol and tobacco consumption history, and family history of cancer, were obtained from patients’ medical records, as listed in Table S1. Tumor type, tumor cell content, histological classification, and cancer grade and stage were reviewed by two pathologists independently. The content of tumor cells in each sample was over 70%, as shown by hematoxylin and eosin staining (Figure S1). The clinical information for the other 46 tumor samples subjected to SNP array genotyping is shown in Table S1. The tumor samples used for SNP array genotyping and mutational rate validation of candidate genes are shown in Figure S2. This study was approved by the Institutional Review Board of the School of Oncology, Peking University, China.
Exome sequencing data processing
Exome sequencing was performed for nine pairs of ESCC tumor samples and the matched blood samples. Genomic DNA libraries were prepared using the Pair-End Genomic DNA Sample Prep Kit (Illumina, San Diego, CA, USA). The genomic DNA was sequenced using the Illumina Genome Analyzer IIx for 75-cycles at BGI, Shenzhen. All original sequencing tags were converted to the FASTAQ format. To increase the accuracy and specificity of read mapping and mutation identification, several pre-processing filters were applied to the raw sequencing tags. For each sequencing tag, if two adjacent bases had a Phred quality score <20, then these two bases and the following bases were trimmed from the tag, and tags shorter than 35 nt were excluded.
Identification and annotation of the somatic mutations
After the pre-processing quality control, tags were aligned to the hg18 version of the human genome using the BWA (Burrows-Wheeler Aligner, version 0.5.8) [57]. Single nucleotide substitutions and small insertion/deletions (indels) were identified using SAMtools (Utilities for the Sequence Alignment/Map format) [58]. Under this filtration, the average number of variants called per sample was ∼15,957 (12,942–21,892), and the transition/transversion (Ti/Tv) ratio was 2.74 (on average, ranging 2.40–2.95 for all samples).
In order to identify and further increase the specificity of somatic mutation calls, we applied the following post-processing filters: (1) only loci with ⩾10× coverage in both tumor and normal samples were used for variant calling; (2) at least 20% of mutant alleles in reads from tumor samples had a Phred quality score ⩾20; and (3) no mutant alleles were detected in reads from the blood samples.
All NSSMs identified by exome sequencing were subjected to Sequenom MassARRAY (Sequenom, San Diego, CA, USA) or conventional Sanger sequencing for validation. Genomic positions and flanking sequences for all SNVs were retrieved using the hg18 version of the human genome and the University of California Santa Cruz (UCSC) genome annotation database. For Sequenom MassARRAY, PCR and MassEXTEND® primers for multiplexed assays were designed using the Sequenom MassARRAY Assay Design 3.1 software. The allele-specific extension products of different masses were quantitatively analyzed using MALDI-TOF mass spectrometry. Mutation calls were determined using a MassARRAY Typer 4.0 Analyzer, according to the manufacturer’s specifications. For Sanger sequencing, PCR primers were designed using Primer Premier 5.0 (PREMIER Biosoft, Palo Alto, CA, USA). The gene ontology (GO) cluster analysis of all mutated genes was done using the Functional Annotation Tool of the Database for Annotation, Visualization and Integrated Discovery (DAVID) [59].
Survey of allelic imbalance
The minor allele fraction (MAF) of each informative SNP was calculated as a measure of allelic imbalance as shown below.
An informative SNP was defined as a known heterozygous SNP at target regions with the coverage of at least 10 distinct reads in both tumor and blood samples. A difference of MAF in blood (MAFB) and tumor (MAFT) samples of more than 0.1 was considered to be significant (MAFB − MAFT ⩾ 0.1). Window sliding was carried out to estimate the size of the altered regions. With a window size of 50 SNPs, a window was considered to be a region undergoing somatic alteration if more than 70% of the SNPs had a significantly-different MAF in the tumor and blood samples.
Whole-genome SNP genotyping and data analysis
Whole-genome SNP array for 55 ESCC samples was performed on Illumina Human OmniZhongHua BeadChips (Illumina, San Diego, CA, USA). Raw intensity values were processed to obtain a normalized B allele frequency (BAF) and a Log R ratio (LRR) for each probe using the Genome Studio Software V2011.1. LRR values were segmented with the Genome Alteration Detection Analysis (GADA) [60] using parameters of T > 10 and segment lengths containing ⩾50 continuous probes. For LOH analysis, the aforementioned window sliding approach was used with a window size of 100 informative SNPs. A window was considered a LOH, if more than 80% of the SNPs had a MAF ⩽ 0.9. A segment was defined either as normal or as having one of 5 types of alteration status based on the following criteria: (1) normal, |LRR| < 0.075 and retaining heterozygosity; (2) gain, LRR ⩾ 0.075; (3) loss, LRR ⩽ −0.075; (4) CNNLOH, |LRR| < 0.075; (5) amplification, LRR ⩾ 0.15; and (6) deletion, LRR ⩽ −0.15.
To survey for genome instability, the genomic fractions of copy number gain, loss, and CNNLOH were estimated by dividing the number of SNPs undergoing a specific alteration by the total number of SNPs present in the respective chromosome or in the respective sample. To identify common regions with copy number gain and loss across samples, the Genomic Identification of Significant Targets in Cancer (GISTIC) algorithm was utilized [61]. Thresholds of LRR were set as 0.15 and −0.15 to allow GISTIC to identify amplifications and deletions, respectively. Q values of MCRs < 0.05 were defined as significant, and 0.99 was used as the confidence level to determine the region that contained potential driver genes.
Statistical analysis
All statistical analysis was performed using R version 3.1.2 or SPSS version 19.0 (IBM, Armonk, New York, USA). The association of overall survival with genomic instability status was evaluated with Kaplan–Meier curves, and differences were tested using the log-rank test. Difference was considered significant with P ⩽ 0.05.
Authors’ contributions
CZ, YK, and HC conceived the study, supervised all aspects of the work, and co-wrote the manuscript. QW and JB performed the analysis and interpretation of sequencing data and SNP array data, participated in majority of the experiments mentioned in the paper, and co-wrote the manuscript. AA, YL, KG, JL, FL, and YP performed sample collection and preparation, somatic mutation validation, and PCR-Sanger sequencing of candidate genes. SL and WS performed SNP genotyping. WC participated in the data analysis of exome sequencing and SNP genotyping. HY, CL, LZ, and RX performed sample collection. All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This study was supported by the National Basic Research Program of China from the National Ministry of Science and Technology (973 Program) to YK (Grant No. 2011CB504300) and to HC (Grant No. 2012CB910800), the National Natural Science Foundation of China (Grant No. 30930102) to YK, the National High-tech R&D Program of China (863 Program; Grant No. 2012AA022502) and Key Research Program of the Chinese Academy of Sciences of China (Grant No. KJZD-EW-L06-2) to CZ, and the Open Fund of MOE Key Laboratory of Carcinogenesis and Translational Research (Grant No. 2014KAIFANG-4) to JB. We would like to thank Chung-I Wu for his suggestions on this manuscript and Michael A. McNutt for editing the manuscript.
Handled by Shaoqi Rao
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
Supplementary material associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.gpb.2015.06.003.
Contributor Information
Hong Cai, Email: drhcai@gmail.com.
Yang Ke, Email: keyang@bjmu.edu.cn.
Changqing Zeng, Email: czeng@big.ac.cn.
Supplementary material
Supplementary Fig. S1.

Hematoxylin and eosin staining of nine tumor samples subjected to exome sequencing. The nine ESCC samples subjected to exome sequencing were examined using hematoxylin and eosin staining. Tumor cell content of each sample (as indicated by patient ID) was evaluated and reviewed by two pathologists independently. The magnification for all of the images was 400×.
Supplementary Fig. S2.
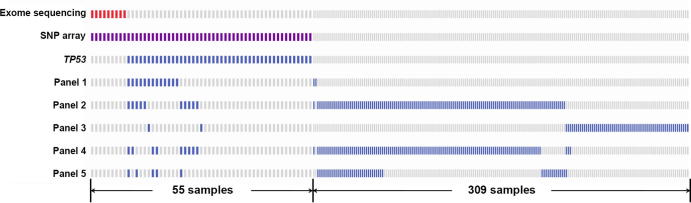
The illustration of 364 samples used in this study. Each small vertical bar in the same column represents an individual case. The bar width of the first 55 samples subjected to SNP assay is twice of that of the remaining 309 samples. Red bars denote the nine samples subjected to exome sequencing, and purple bars denote the 55 samples subjected to whole-genome SNP array. Samples tested by Sanger sequencing for the verification of somatic mutation in TP53 were represented in blue bars. Sanger sequencing panels are defined as: Panel 1: CHD3, MLL, NASP, PHF16, UBN2; Panel 2: CD40LG, FBXL4, INHBC, FBXW7, ANG; Panel 3: WNT2B, TSPYL2, SMARCD3; Panel 4: BRAF, AKT1, PIK3CA; and Panel 5: XRCC2. Colored bars indicate the samples that were enrolled in the specific sequencing or array panels, while grey bars indicate the samples that were not included. Samples enrolled in each panel were picked randomly.
Supplementary Fig. S3.

Genes with somatic point mutations in ESCC patients identified by exome sequencing. Only TP53 mutations were seen in five samples, while FBXL4 and DMD had recurrent mutations recovered in 2 samples.
Supplementary Fig. S4.

Circular plot showing 107 imbalanced allelic events in autosomes identified by exome sequencing. The outer ring shows autosomes 1–22 with the respective centromeres marked in red. Events and positions of allelic imbalance on each autosome are indicated by the blue bars in the grey inner ring. Blue bars were stacked up when alteration events occurred at overlapping genomic positions.
Supplementary Fig. S5.

Low genomic alterations in nine blood samples from ESCC patients identified by using cnvPartition. Across autosomes 1−22, regions with CNAs are indicated with colored bars as follows: regions with copy number = 0–0.5 (red), 0.5–1.5 (orange), 1.5–2.5 (CNNLOH, green), 2.5–3.5 (blue), and 3.5–4.5 (purple). Patient ID was listed below each sample. CNA, copy number alteration.
Supplementary Fig. S6.

Overview of genomic alteration fractions on chromosome arms across 55 ESCC samples. CNG, copy number gain; CNL, copy number loss; CNNLOH, copy number neutral loss of heterozygosity.
Supplementary Fig. S7.

Association between genomic instability level and patient survival rate. Patients with higher degrees of genomic instability (⩾50% of the genome showing alterations, 25 patients) tended to have poorer post-surgery survival than those with a low degree of genomic alteration (<50% of the genome showed alterations, 24 patients), though the difference was not statistically significant (P = 0.083).
Clinicopathologic information of ESCC patients subjected to exome sequencing and SNP array genotyping.
Regions of focal amplification and deletion and the genes harbored.
Correlation between genomic instability and clinical data.
Minimal common regions associated with lymph node metastases.
The average read depths and fraction of coverage under different read depths of exome sequencing for each sample.
Characterization of non-synonymous somatic mutations and indels identified in the nine patients subjected to exome sequencing.
TP53 mutations identified in 23/46 samples by Sanger sequencing.
Top five GO biological processes enriched.
Fractions of genomic alterations in 55 ESCCs detected by whole-genome SNP array.
Fractions of genomic alterations in nine blood samples from ESCC patients detected by whole-genome SNP array.
Fractions of genomic alterations in blood samples detected using cnvPartition.
Fractions of genomic alterations on different autosomal arms in 55 ESCC tumor samples.
References
- 1.Torre L.A., Bray F., Siegel R.L., Ferlay J., Lortet-Tieulent J., Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Hu J., Nyren O., Wolk A., Bergstrom R., Yuen J., Adami H.O. Risk factors for oesophageal cancer in Northeast China. Int J Cancer. 1994;57:38–46. doi: 10.1002/ijc.2910570108. [DOI] [PubMed] [Google Scholar]
- 4.Enzinger P.C., Mayer R.J. Esophageal cancer. N Engl J Med. 2003;349:2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 5.Mariette C., Balon J.M., Piessen G., Fabre S., Van Seuningen I., Triboulet J.P. Pattern of recurrence following complete resection of esophageal carcinoma and factors predictive of recurrent disease. Cancer. 2003;97:1616–1623. doi: 10.1002/cncr.11228. [DOI] [PubMed] [Google Scholar]
- 6.Sant M., Aareleid T., Berrino F., Bielska Lasota M., Carli P.M., Faivre J. EUROCARE-3: survival of cancer patients diagnosed 1990–94 – results and commentary. Ann Oncol. 2003;14 Suppl 5:v61–v118. doi: 10.1093/annonc/mdg754. [DOI] [PubMed] [Google Scholar]
- 7.Yuequan J., Shifeng C., Bing Z. Prognostic factors and family history for survival of esophageal squamous cell carcinoma patients after surgery. Ann Thorac Surg. 2010;90:908–913. doi: 10.1016/j.athoracsur.2010.05.060. [DOI] [PubMed] [Google Scholar]
- 8.Kajiyama Y., Kanno H., Ueno M., Udagawa H., Tsutsumi K., Kinoshita Y. P53 gene mutation in 150 dissected lymph nodes in a patient with esophageal cancer. Dis Esophagus. 1998;11:279–283. doi: 10.1093/dote/11.4.279. [DOI] [PubMed] [Google Scholar]
- 9.Wang X.L., Zhang C.M., Shi L.Y., Yu H.P., Xu S.Q. Significance of p53 gene mutation and P53 protein expression abnormality on the prognosis of esophageal cancer: a meta-analysis study. Zhonghua Liu Xing Bing Xue Za Zhi. 2004;25:769–774. [PubMed] [Google Scholar]
- 10.Cai Y.C., So C.K., Nie A.Y., Song Y., Yang G.Y., Wang L.D. Characterization of genetic alteration patterns in human esophageal squamous cell carcinoma using selected microsatellite markers spanning multiple loci. Int J Oncol. 2007;30:1059–1067. [PubMed] [Google Scholar]
- 11.Wiech T., Nikolopoulos E., Weis R., Langer R., Bartholome K., Timmer J. Genome-wide analysis of genetic alterations in Barrett’s adenocarcinoma using single nucleotide polymorphism arrays. Lab Invest. 2009;89:385–397. doi: 10.1038/labinvest.2008.67. [DOI] [PubMed] [Google Scholar]
- 12.Hu N., Clifford R.J., Yang H.H., Wang C., Goldstein A.M., Ding T. Genome wide analysis of DNA copy number neutral loss of heterozygosity (CNNLOH) and its relation to gene expression in esophageal squamous cell carcinoma. BMC Genomics. 2010;11:576. doi: 10.1186/1471-2164-11-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makino T., Yamasaki M., Miyata H., Yoshioka S., Takiguchi S., Fujiwara Y. P53 mutation status predicts pathological response to chemoradiotherapy in locally advanced esophageal cancer. Ann Surg Oncol. 2010;17:804–811. doi: 10.1245/s10434-009-0786-9. [DOI] [PubMed] [Google Scholar]
- 14.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiegand K.C., Shah S.P., Al-Agha O.M., Zhao Y., Tse K., Zeng T. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le Gallo M., O’Hara A.J., Rudd M.L., Urick M.E., Hansen N.F., O’Neil N.J. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44:1310–1315. doi: 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones S., Wang T.L., Shih Ie M., Mao T.L., Nakayama K., Roden R. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zang Z.J., Cutcutache I., Poon S.L., Zhang S.L., McPherson J.R., Tao J. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570–574. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 19.Varela I., Tarpey P., Raine K., Huang D., Ong C.K., Stephens P. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konsavage W.M., Jr., Kyler S.L., Rennoll S.A., Jin G., Yochum G.S. Wnt/beta-catenin signaling regulates Yes-associated protein (YAP) gene expression in colorectal carcinoma cells. J Biol Chem. 2012;287:11730–11739. doi: 10.1074/jbc.M111.327767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinhardt A.A., Gayyed M.F., Klein A.P., Dong J., Maitra A., Pan D. Expression of Yes-associated protein in common solid tumors. Hum Pathol. 2008;39:1582–1589. doi: 10.1016/j.humpath.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zender L., Spector M.S., Xue W., Flemming P., Cordon-Cardo C., Silke J. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–1267. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A., Shendure J., Nelson P.S. Genome interrupted: sequencing of prostate cancer reveals the importance of chromosomal rearrangements. Genome Med. 2011;3:23. doi: 10.1186/gm237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berger M.F., Lawrence M.S., Demichelis F., Drier Y., Cibulskis K., Sivachenko A.Y. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stephens P.J., Greenman C.D., Fu B., Yang F., Bignell G.R., Mudie L.J. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao Y.B., Chen Z.L., Li J.G., Hu X.D., Shi X.J., Sun Z.M. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097–1102. doi: 10.1038/ng.3076. [DOI] [PubMed] [Google Scholar]
- 27.Lin D.C., Hao J.J., Nagata Y., Xu L., Shang L., Meng X. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46:467–473. doi: 10.1038/ng.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song Y., Li L., Ou Y., Gao Z., Li E., Li X. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91–95. doi: 10.1038/nature13176. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L., Zhou Y., Cheng C., Cui H., Cheng L., Kong P. Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. Am J Hum Genet. 2015;96:597–611. doi: 10.1016/j.ajhg.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Livingstone L.R., White A., Sprouse J., Livanos E., Jacks T., Tlsty T.D. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 1992;70:923–935. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- 31.Murray A.W. The genetics of cell cycle checkpoints. Curr Opin Genet Dev. 1995;5:5–11. doi: 10.1016/s0959-437x(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 32.Elledge S.J. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 33.Negrini S., Gorgoulis V.G., Halazonetis T.D. Genomic instability – an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 34.Elsheikh S., Green A.R., Aleskandarany M.A., Grainge M., Paish C.E., Lambros M.B. CCND1 amplification and cyclin D1 expression in breast cancer and their relation with proteomic subgroups and patient outcome. Breast Cancer Res Treat. 2008;109:325–335. doi: 10.1007/s10549-007-9659-8. [DOI] [PubMed] [Google Scholar]
- 35.Foulkes W.D., Flanders T.Y., Pollock P.M., Hayward N.K. The CDKN2A (p16) gene and human cancer. Mol Med. 1997;3:5–20. [PMC free article] [PubMed] [Google Scholar]
- 36.Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 37.Audrezet M.P., Robaszkiewicz M., Mercier B., Nousbaum J.B., Bail J.P., Hardy E. TP53 gene mutation profile in esophageal squamous cell carcinomas. Cancer Res. 1993;53:5745–5749. [PubMed] [Google Scholar]
- 38.Qin Y.R., Wang L.D., Fan Z.M., Kwong D., Guan X.Y. Comparative genomic hybridization analysis of genetic aberrations associated with development of esophageal squamous cell carcinoma in Henan, China. World J Gastroenterol. 2008;14:1828–1835. doi: 10.3748/wjg.14.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu N., Wang C., Hu Y., Yang H.H., Kong L.H., Lu N. Genome-wide loss of heterozygosity and copy number alteration in esophageal squamous cell carcinoma using the Affymetrix GeneChip Mapping 10 K array. BMC Genomics. 2006;7:299. doi: 10.1186/1471-2164-7-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hao J.J., Gong T., Zhang Y., Shi Z.Z., Xu X., Dong J.T. Characterization of gene rearrangements resulted from genomic structural aberrations in human esophageal squamous cell carcinoma KYSE150 cells. Gene. 2013;513:196–201. doi: 10.1016/j.gene.2012.09.091. [DOI] [PubMed] [Google Scholar]
- 41.Hu N., Wang C., Ng D., Clifford R., Yang H.H., Tang Z.Z. Genomic characterization of esophageal squamous cell carcinoma from a high-risk population in China. Cancer Res. 2009;69:5908–5917. doi: 10.1158/0008-5472.CAN-08-4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guichard C., Amaddeo G., Imbeaud S., Ladeiro Y., Pelletier L., Maad I.B. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jong Y.J., Li L.H., Tsou M.H., Chen Y.J., Cheng S.H., Wang-Wuu S. Chromosomal comparative genomic hybridization abnormalities in early- and late-onset human breast cancers: correlation with disease progression and TP53 mutations. Cancer Genet Cytogenet. 2004;148:55–65. doi: 10.1016/s0165-4608(03)00205-x. [DOI] [PubMed] [Google Scholar]
- 44.Eyfjord J.E., Thorlacius S., Steinarsdottir M., Valgardsdottir R., Ogmundsdottir H.M., Anamthawat-Jonsson K. P53 abnormalities and genomic instability in primary human breast carcinomas. Cancer Res. 1995;55:646–651. [PubMed] [Google Scholar]
- 45.Overholtzer M., Rao P.H., Favis R., Lu X.Y., Elowitz M.B., Barany F. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc Natl Acad Sci U S A. 2003;100:11547–11552. doi: 10.1073/pnas.1934852100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gorgoulis V.G., Vassiliou L.V., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 47.Bartkova J., Horejsi Z., Koed K., Kramer A., Tort F., Zieger K. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 48.Di Micco R., Fumagalli M., Cicalese A., Piccinin S., Gasparini P., Luise C. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 49.Di Agostino S., Strano S., Emiliozzi V., Zerbini V., Mottolese M., Sacchi A. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10:191–202. doi: 10.1016/j.ccr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 50.Ozaki T., Nakagawara A. P53: the attractive tumor suppressor in the cancer research field. J Biomed Biotechnol. 2011;2011:603925. doi: 10.1155/2011/603925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGivern D.R., Lemon S.M. Tumor suppressors, chromosomal instability, and hepatitis C virus-associated liver cancer. Annu Rev Pathol. 2009;4:399–415. doi: 10.1146/annurev.pathol.4.110807.092202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Janssen J.W., Vaandrager J.W., Heuser T., Jauch A., Kluin P.M., Geelen E. Concurrent activation of a novel putative transforming gene, myeov, and cyclin D1 in a subset of multiple myeloma cell lines with t(11;14)(q13;q32) Blood. 2000;95:2691–2698. [PubMed] [Google Scholar]
- 53.Zhai C., Li Y., Mascarenhas C., Lin Q., Li K., Vyrides I. The function of ORAOV1/LTO1, a gene that is overexpressed frequently in cancer: essential roles in the function and biogenesis of the ribosome. Oncogene. 2014;33:484–494. doi: 10.1038/onc.2012.604. [DOI] [PubMed] [Google Scholar]
- 54.Ruiz C., Martins J.R., Rudin F., Schneider S., Dietsche T., Fischer C.A. Enhanced expression of ANO1 in head and neck squamous cell carcinoma causes cell migration and correlates with poor prognosis. PLoS One. 2012;7:e43265. doi: 10.1371/journal.pone.0043265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dedes K.J., Lopez-Garcia M.A., Geyer F.C., Lambros M.B., Savage K., Vatcheva R. Cortactin gene amplification and expression in breast cancer: a chromogenic in situ hybridisation and immunohistochemical study. Breast Cancer Res Treat. 2010;124:653–666. doi: 10.1007/s10549-010-0816-0. [DOI] [PubMed] [Google Scholar]
- 56.Fantozzi I., Grall D., Cagnol S., Stanchi F., Sudaka A., Brunstein M.C. Overexpression of cortactin in head and neck squamous cell carcinomas can be uncoupled from augmented EGF receptor expression. Acta Oncol. 2008;47:1502–1512. doi: 10.1080/02841860802089801. [DOI] [PubMed] [Google Scholar]
- 57.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pique-Regi R., Caceres A., Gonzalez J.R. R-Gada: a fast and flexible pipeline for copy number analysis in association studies. BMC Bioinformatics. 2010;11:380. doi: 10.1186/1471-2105-11-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinicopathologic information of ESCC patients subjected to exome sequencing and SNP array genotyping.
Regions of focal amplification and deletion and the genes harbored.
Correlation between genomic instability and clinical data.
Minimal common regions associated with lymph node metastases.
The average read depths and fraction of coverage under different read depths of exome sequencing for each sample.
Characterization of non-synonymous somatic mutations and indels identified in the nine patients subjected to exome sequencing.
TP53 mutations identified in 23/46 samples by Sanger sequencing.
Top five GO biological processes enriched.
Fractions of genomic alterations in 55 ESCCs detected by whole-genome SNP array.
Fractions of genomic alterations in nine blood samples from ESCC patients detected by whole-genome SNP array.
Fractions of genomic alterations in blood samples detected using cnvPartition.
Fractions of genomic alterations on different autosomal arms in 55 ESCC tumor samples.