

ABSTRACT
Insensitivity and technical complexity have impeded the implementation of high-throughput nucleic acid sequencing in differential diagnosis of viral infections in clinical laboratories. Here, we describe the development of a virome capture sequencing platform for vertebrate viruses (VirCapSeq-VERT) that increases the sensitivity of sequence-based virus detection and characterization. The system uses ~2 million probes that cover the genomes of members of the 207 viral taxa known to infect vertebrates, including humans. A biotinylated oligonucleotide library was synthesized on the NimbleGen cleavable array platform and used for solution-based capture of viral nucleic acids present in complex samples containing variable proportions of viral and host nucleic acids. The use of VirCapSeq-VERT resulted in a 100- to 10,000-fold increase in viral reads from blood and tissue homogenates compared to conventional Illumina sequencing using established virus enrichment procedures, including filtration, nuclease treatments, and RiboZero rRNA subtraction. VirCapSeq-VERT had a limit of detection comparable to that of agent-specific real-time PCR in serum, blood, and tissue extracts. Furthermore, the method identified novel viruses whose genomes were approximately 40% different from the known virus genomes used for designing the probe library. The VirCapSeq-VERT platform is ideally suited for analyses of virome composition and dynamics.
Importance VirCapSeq-VERT enables detection of viral sequences in complex sample backgrounds, including those found in clinical specimens, such as serum, blood, and tissue. The highly multiplexed nature of the system allows both the simultaneous identification and the comprehensive genetic characterization of all known vertebrate viruses, their genetic variants, and novel viruses. The operational simplicity and efficiency of the VirCapSeq-VERT platform may facilitate transition of high-throughput sequencing to clinical diagnostic as well as research applications.
Importance
VirCapSeq-VERT enables detection of viral sequences in complex sample backgrounds, including those found in clinical specimens, such as serum, blood, and tissue. The highly multiplexed nature of the system allows both the simultaneous identification and the comprehensive genetic characterization of all known vertebrate viruses, their genetic variants, and novel viruses. The operational simplicity and efficiency of the VirCapSeq-VERT platform may facilitate transition of high-throughput sequencing to clinical diagnostic as well as research applications.
INTRODUCTION
Clinical virology and virus discovery in the 20th century focused chiefly on the identification of viruses through microscopy, serology, and cell or animal infection studies (1). With the advent of nucleic acid amplification, a wide range of molecular approaches for virus detection became available: PCR (2), consensus PCR (cPCR) and multiplex PCR systems (3–10), differential display (11), representational difference analysis (12, 13), subtractive cloning (14), domain-specific differential display (15), cDNA cloning (16–18), cDNA immunoscreening (19, 20), microarrays (21, 22), and, most recently, high-throughput sequencing (HTS). HTS has enabled unbiased pathogen discovery and facilitated virome analyses that have enhanced our understanding of the origin, evolution, and ecology of known and novel viruses (1). However, HTS has not been widely implemented in clinical diagnostic laboratories largely due to operational complexity, cost, and insensitivity with respect to agent-specific PCR assays.
Strategies to increase the sensitivity of HTS have focused on the enrichment of viral template through subtraction of host nucleic acid via nuclease digestion and depletion of rRNA. Although they are helpful, none has achieved the sensitivity required for clinical applications. To address this challenge, we have established a positive selection probe capture-based system to enrich sequence libraries for viral sequences. Here, we describe the virome capture sequencing platform for vertebrate viruses (VirCapSeq-VERT) and demonstrate its potential utility as a sensitive and specific HTS-based platform for clinical diagnosis and virome analysis.
RESULTS
Probe design strategy.
Our objective was to target all known viruses that can infect vertebrate animals, including humans. Toward this end, oligonucleotides were selected to represent all viral taxa containing at least one virus known to infect vertebrates; virus families that include exclusively viruses infecting plants or insects were excluded (see Table S1 in the supplemental material). Coding sequences were extracted from the EMBL Coding Domain Sequence database, clustered at 96% sequence identity, and used to select 100-mer oligonucleotides spaced by approximately 25 to 50 nucleotides (nt) along each sequence. To address sequence variation, oligonucleotide mutant or variant sequences were retained if sequences diverged by more than 90%. Where technical complexity in oligonucleotide synthesis was challenging due to melting temperature (Tm) or homopolymer repeats, probe sequences were refined by shortening and adjusting their start/stop positions. The final library comprised 1,993,176 oligonucleotides ranging in length from 50 to 100 nt and in Tm from 58.7°C to 101°C (see Table S2 in the supplemental material).
We evaluated in silico whether the selected probe library provides uniform coverage of the targeted virus sequences. Our analysis indicated that probe numbers were proportional to the amount of available sequence information, resulting in an 88 to 98% estimated coverage of target sequences when an “outreach” for each probe of approximately 100 nt to either side is assumed (see Table S3 in the supplemental material). We mapped the probe library against a database of 100 reference virus genome sequences representing double- and single-stranded DNA and RNA, positive and negative RNA, and circular, linear, and segmented viruses, using a minimum nucleotide identity of 90%. The probe library covered targeted genome sequences with probes spaced at <150-nt intervals (Fig. 1) but provided no coverage of noncoding regions (e.g., poliovirus 5′ untranslated region [UTR]) (Fig. 1A). The highest probe coverage was evident in divergent genome regions (e.g., yellow fever virus E gene region; approximately position 1000 to 2500) (Fig. 1B). In silico analysis indicated that the VirCapSeq-VERT probe library included oligonucleotides that selectively hybridize to genomes of vertebrate viruses but not to those of bacteriophages or plant or fungal viruses.
FIG 1 .

In silico validation of the VirCapSeq-VERT probe design. Probe depth and coverage of the VirCapSeq-VERT probe library are shown for poliovirus (A), yellow fever virus (B), and parvovirus B19 (C). Virus genomes are represented by black lines. The coding sequences are represented by green boxes. The probes are indicated by grey boxes. The top graph in each panel indicates probe depth at each locus. Colored lines in the probes indicate mismatch to the reference used for alignment (green, A; red, T; blue, C; orange, G). Line heights in the coverage track above indicate frequency of the mismatched bases.
Experimental assessment of efficiency.
Nucleic acid (NA) extracts of human lung tissue or whole blood were spiked with various amounts of NAs representing large and small, positive- and negative-strand, segmented and nonsegmented, and DNA and RNA viruses (Table 1). Spiked lung (pool 1) and blood (pool 2) NA preparations were divided and processed in parallel using a standard Illumina HTS protocol or the VirCapSeq-VERT system, whereby viral sequences are enriched by positive selection. Each of the preparations was sequenced on an Illumina HiSeq 2500 sequencer, loading 2 lanes per pool. VirCapSeq-VERT resulted in a 100- to 1,000-fold increase in on-target (viral) reads and a reduction of host background reads from 99.7% to 68.2% in lung and from 99.4% to 38.5% in blood (Fig. 2; also, see Table S4 in the supplemental material). The average coverage also increased dramatically, with nearly full-length sequences (>95%) being obtained for all viruses (Table 2). Figure 3 shows selected examples of sequence recovery for West Nile virus (WNV), Cache Valley virus (CVV), and Middle East respiratory syndrome coronavirus (MERS-CoV).
TABLE 1 .
Assessment of VirCapSeq-VERT efficiency by using quantitated viral nucleic acids to spike lung and blood host nucleic acid
| Pool | Host background | Virusb | Loada | Library preparation |
|---|---|---|---|---|
| 1a | 200 ng lung NA | FLUAV (Orthomyxoviridae); segmented negative-strand RNA, 13 kb/8 segments |
2 × 104 | Conventional/HTS |
| MERS-CoV (Nidovirales, Coronaviridae); nonsegmented positive-strand RNA, 30 kb |
2 × 104 | |||
| EV-D68 (Picornavirales, Picornaviridae); nonsegmented positive-strand RNA, 7 kb |
3 × 105 | |||
| 1b | Same as pool 1a | Same as pool 1a | Same as pool 1a | VirCapSeq-VERT |
| 2a | 200 ng blood NA | DENV-3 (Flaviviridae); nonsegmented positive-strand RNA, 11 kb |
5 × 105 | Conventional/HTS |
| WNV (Flaviviridae); nonsegmented positive-strand RNA, 11 kb |
9 × 103 | |||
| EBOV (Mononegavirales, Filoviridae); nonsegmented negative-strand RNA, 19 kb |
2 × 103 | |||
| CVV (Bunyaviridae); segmented negative-strand RNA, 12 kb/3 segments |
8 × 103 | |||
| HHV-1 (Herpesvirales, Herpesviridae); nonsegmented double-strand DNA, 152 kb |
2 × 105 | |||
| 2b | Same as pool 2a | Same as pool 2a | Same as pool 2a | VirCapSeq-VERT |
Determined by qPCR of double-stranded cDNA/DNA used for sequence library construction.
FLUAV, influenza A virus H3N2; MERS-CoV, Middle East respiratory syndrome coronavirus; EV-D68, enterovirus D68; DENV-3, dengue virus 3; WNV, West Nile virus; EBOV, Ebola virus; CVV, Cache Valley virus; HHV-1, herpes simplex virus 1.
FIG 2 .

VirCapSeq-VERT enhances the performance of high-throughput sequencing by increasing the number of mapped viral reads recovered from high-background specimens. Eight different viral NAs were quantitated by qPCR and used to spike a background of lung-derived (3 viruses) or blood-derived (5 viruses) NA extracts. Samples were split in two and processed by standard HTS (blue) or with VirCapSeq-VERT (red). FLUAV, influenza A virus; EVD-68, enterovirus D68; MERS-CoV, MERS coronavirus; DENV, dengue virus; EBOV, Ebola virus; WNV, West Nile virus; CVV, Cache Valley virus; HHV-1, human herpesvirus 1.
TABLE 2 .
VirCapSeq-VERT provides greater genome coverage and sequencing depth than HTS
| Libraryb | Virus | Load (copies)a | Genome length (nt) | No. of mapped positions | % sequence mapped | Coverage |
No. of unmapped regions | Unmapped region length (nt) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Min | Max | Avg | ||||||||
| Pool 1a (lung, HTS) | EV-D68 | 105 | 7,341 | 7,268 | 99.01 | 0 | 2,384 | 932 | 4 | 73 |
| MERS-CoV | 104 | 30,113 | 1,824 | 6.06 | 0 | 2 | 0.1 | 19 | 28,289 | |
| FLUAV -1 | 104 | 2,316 | 2,005 | 86.57 | 0 | 9 | 2.5 | 5 | 311 | |
| FLUAV -2 | 2,304 | 2,248 | 97.57 | 0 | 19 | 6.4 | 2 | 56 | ||
| FLUAV -3 | 2,208 | 1,998 | 90.49 | 0 | 29 | 3.8 | 4 | 210 | ||
| FLUAV -4 | 1,737 | 1,642 | 94.53 | 0 | 32 | 8.0 | 2 | 95 | ||
| FLUAV -5 | 1,540 | 1,494 | 97.01 | 0 | 14 | 4.1 | 3 | 46 | ||
| FLUAV -6 | 1,442 | 1,334 | 92.51 | 0 | 11 | 4.2 | 3 | 108 | ||
| FLUAV -7 | 1,002 | 948 | 94.61 | 0 | 11 | 3.7 | 2 | 54 | ||
| FLUAV -8 | 865 | 801 | 92.60 | 0 | 11 | 3.8 | 3 | 65 | ||
| Pool 1b (lung, VirCapSeq-VERT) | EV-D68 | 105 | 7,341 | 7,341 | 100.00 | 3 | 8,080 | 7,005 | 0 | 0 |
| MERS-CoV | 104 | 30,113 | 29,020 | 96.37 | 0 | 121 | 13 | 23 | 1,093 | |
| FLUAV -1 | 104 | 2,316 | 2,316 | 100.00 | 590 | 8,061 | 5,230 | 0 | 0 | |
| FLUAV -2 | 2,304 | 2,304 | 100.00 | 569 | 8,048 | 7,608 | 0 | 0 | ||
| FLUAV -3 | 2,208 | 2,208 | 100.00 | 818 | 8,040 | 4,847 | 0 | 0 | ||
| FLUAV -4 | 1,737 | 1,737 | 100.00 | 323 | 8,038 | 7,449 | 0 | 0 | ||
| FLUAV -5 | 1,540 | 1,540 | 100.00 | 909 | 8,003 | 7,091 | 0 | 0 | ||
| FLUAV -6 | 1,442 | 1,442 | 100.00 | 348 | 7,999 | 6,975 | 0 | 0 | ||
| FLUAV -7 | 1,002 | 1,002 | 100.00 | 60 | 8,056 | 6,216 | 0 | 0 | ||
| FLUAV -8 | 865 | 865 | 100.00 | 448 | 8,006 | 5,761 | 0 | 0 | ||
| Pool 2a (blood, HTS) | HHV-1 | 105 | 152,151 | 151,970 | 99.88 | 0 | 418 | 142 | 4 | 183 |
| DENV-3 | 105 | 10,707 | 10,687 | 99.81 | 0 | 1,242 | 622 | 1 | 20 | |
| WNV | 104 | 10,945 | 500 | 4.57 | 0 | 1 | 0.1 | 6 | 10,445 | |
| EBOV | 103 | 18,959 | 4,716 | 24.87 | 0 | 2 | 0.3 | 43 | 14,243 | |
| CVV-S | 104 | 905 | 818 | 90.39 | 0 | 7 | 3.1 | 3 | 87 | |
| CVV-M | 4,305 | 2,633 | 61.16 | 0 | 5 | 1.1 | 15 | 1,672 | ||
| CVV-L | 6,840 | 2,309 | 33.79 | 0 | 5 | 0.5 | 17 | 4,531 | ||
| Pool 2b (blood, VirCapSeq-VERT) | HHV-1 | 105 | 152,151 | 152,133 | 99.99 | 0 | 8,001 | 5,373 | 1 | 18 |
| DENV-3 | 105 | 10,707 | 10,688 | 99.82 | 0 | 8,068 | 7,774 | 1 | 19 | |
| WNV | 104 | 10,945 | 10,428 | 95.28 | 0 | 214 | 66 | 1 | 517 | |
| EBOV | 103 | 18,959 | 16,413 | 86.57 | 0 | 394 | 56 | 11 | 2,546 | |
| CVV-S | 104 | 905 | 904 | 99.89 | 0 | 7,319 | 2,302 | 1 | 1 | |
| CVV-M | 4,305 | 4,305 | 100.00 | 2 | 1,551 | 401 | 0 | 0 | ||
| CVV-L | 6,840 | 6,840 | 100.00 | 1 | 858 | 88 | 0 | 0 | ||
Determined by qPCR of double-stranded-cDNA/DNA used for sequence library construction.
See Table 1 for pool composition.
FIG 3 .

Read coverage versus probe coverage of VirCapSeq-VERT for West Nile virus (A), Cache Valley virus (B), and MERS coronavirus (C). Virus genomes are represented by horizontal black lines and coding sequence by black pointed boxes. The top graph in each panel indicates the read coverage obtained by VirCapSeq-VERT; probe coverage is shown below. Colored lines indicate mismatch to the reference used for alignment (green, A; red, T; blue, C; orange, G). Line heights indicate the frequency of the mismatched bases.
To determine the threshold for detection of viral sequence, we used NA from lung tissue homogenate and EDTA-blood that contained different amounts of WNV and herpes simplex virus 1 (HHV-1) NA. Nearly complete genome recovery (>90%) was achieved for both viruses at input levels of 100 viral copies in 50 ng of blood NA or 1,000 viral copies in 100 ng lung NA (Fig. 4). Extrapolated to clinical samples, these values correspond to a blood specimen containing approximately 1,200 copies/ml or a tissue specimen containing approximately 16,000 copies/mg, assuming 100% extraction yield (see Table S5 in the supplemental material). Even at the lowest level of virus input tested, 10 viral copies per 50 ng background NA, corresponding to approximately 100 copies/ml blood, VirCapSeq-VERT enabled recovery of 45 kb (29%) of HHV-1 and 0.75 kb (7%) of WNV genome sequence. We then tested human blood and serum samples (1 ml) spiked with live enterovirus D68 (EV-D68) virus stock. VirCapSeq-VERT enabled detection in both sample types at a concentration of 10 copies/ml (Fig. 5), comparable to the sensitivity of real-time PCR (see Table S6 in the supplemental material).
FIG 4 .
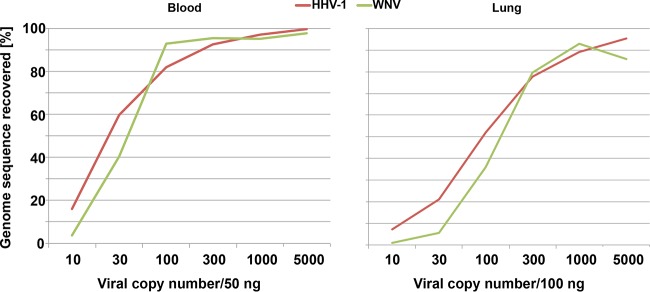
Limit of detection for VirCapSeq-VERT. Total nucleic acid from blood or lung tissue was spiked with human herpesvirus 1 (HHV-1) and West Nile virus (WNV) nucleic acid. The two preparations were serially diluted to generate six samples containing both viruses at 5,000, 1,000, 300, 100, 30, or 10 copies in 100 ng lung tissue or 50 ng whole-blood nucleic acid. Samples were processed with VirCapSeq-VERT.
FIG 5 .
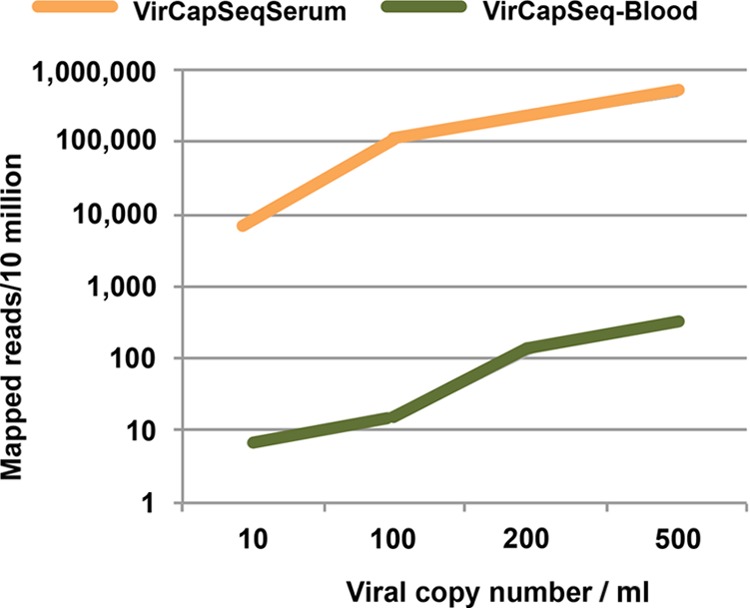
Efficiency of viral read mapping with VirCapSeq-VERT. Human blood and serum were spiked with live enterovirus D68 virus stock quantitated by qPCR to generate samples with 500, 200, 100, or 10 copies/ml. Five hundred microliters of each sample was extracted and processed with VirCapSeq-VERT.
Comparison with other enrichment regimens.
Analysis of samples of human blood spiked with live EV-D68, HHV-1, and influenza A virus (FLUAV) stock indicated that VirCapSeq-VERT yielded up to a 10,000-fold increase in mapped read counts over samples treated after extraction with DNase and RiboZero rRNA depletion, individually or in combination, and then processed by standard HTS. VirCapSeq-VERT resulted in nearly full genome recovery for most viruses even with less than 1,000 copies of target input (Table 3; also, see Table S7 in the supplemental material).
TABLE 3 .
VirCapSeq-VERT performance compared to conventional enrichment procedures
| Treatment (preparation)a | Virus load (copies)b (HHV/FLUAV/EV) | No. of reads | No. of reads (total/normalizedd) |
|||
|---|---|---|---|---|---|---|
| Viral | Mapped to virus |
|||||
| HHV-1 | FLUAV | EV-D68 | ||||
| DNase (conventional) | 6 × 102/ND/9 × 102 | 20,449,329 | 219/107 | 59/29c | 6/3 | 154/75 |
| RiboZero (conventional) | 2 × 103/8 × 102/2 × 103 | 82,866,269 | 4,251/513 | 2,951/356 | 39/5 | 1,261/152 |
| DNase/RiboZero (conventional) | ND/ND/2 × 103 | 68,239,834 | 3,927/576 | 6/0.9c | 3/0.4 | 3,918/575 |
| None (conventional) | 2 × 104/3 × 104/2 × 104 | 121,961,881 | 4,562/374 | 2,569/211 | 65/5 | 1,928/158 |
| None (VirCapSeq-VERT) | 2 × 104/2 × 104/2 × 104 | 128,764,130 | 2,773,382/215,325 | 713,557/55,400 | 572,169/44,423 | 1,487,656/115,501 |
| None (VirCapSeq-VERT)e | 9 × 102/8 × 102/9 × 102 | 64,989,060 | 86,943/13,376 | 21,631/3,328 | 19,255/2,962 | 46,057/7,086 |
Human blood was spiked with live virus stocks derived from tissue culture to result in approximately 104 copies of herpes simplex virus 1 (HHV-1), influenza A virus (FLUAV), and enterovirus D68 (EV-D68) per 250 ng extracted blood NA. The sample was divided into equivalent aliquots to be processed with the indicated treatment prior to RT reaction and subjected to either conventional sequence library preparation or VirCapSeq-VERT.
Determined by qPCR of double-stranded cDNA/DNA used for sequence library construction.
HHV-1 detection was impaired due to DNase.
Normalized to 10,000,000 total reads.
Prepared with additional dilution of the sample in a blood background.
Clinical specimens included a human nasal swab sample containing EV-D68 that was divided into three aliquots (i) treated with filtration and nuclease digestion prior to extraction and standard HTS, (ii) treated with filtration and nuclease digestion prior to extraction and VirCapSeq-VERT, or (iii) not treated prior to extraction and VirCapSeq-VERT. VirCapSeq-VERT with no prior treatment enabled the highest sequence recovery and depth (see Table S8 in the supplemental material).
Since fecal material is frequently challenging for viromic analyses, we tested a sample of fecal pellets from bats known to contain rotavirus sequences. The sample was divided into four aliquots and (i) treated with filtration and nuclease digestion prior to extraction, followed by standard HTS, (ii) treated with filtration and nuclease digest prior to extraction, followed by DNase digestion after extraction and standard HTS, (iii) treated with filtration and nuclease digestion prior to extraction and VirCapSeq-VERT, or (iv) not treated prior to extraction and VirCapSeq-VERT. VirCapSeq-VERT again yielded the highest mapped read count (see Table S9 in the supplemental material).
The specificity of VirCapSeq-VERT for relevant targets was readily apparent in comparison with results obtained by conventional HTS. Whereas up to 36% of the viral reads found by conventional HTS represented insect-infecting dicistroviruses (Fig. 6), these reads were reduced to 15% by VirCapSeq-VERT. Vertebrate rotavirus, coronavirus, astrovirus, and circovirus sequences detected only at low levels in conventional HTS were increased 4-100 fold by VirCapSeq-VERT.
FIG 6 .

Selective enhancement of vertebrate virus detection by VirCapSeq-VERT. Bat fecal sample material was divided in two and analyzed using HTS with filtration and nuclease digest combined with postextraction DNase treatment or using VirCapSeq-VERT alone. VirCapSeq-VERT reduced the number of nonvertebrate viral reads and efficiently sequenced vertebrate virus sequences detected only at low levels by conventional HTS.
Since the precise sequence of the rotavirus present in bat samples was unknown, contigs obtained by de novo assembly were used to identify the closest GenBank match for each segment. Table 4 shows that nearly a full sequence was obtained for sequences differing by up to 25% from the known sequences used for VirCapSeq-VERT probe design. Partial sequence in conserved regions was obtained even for sequences differing by as much as 50% from known sequences (NSP1 and NSP4) (Table 4).
TABLE 4 .
Capacity of VirCapSeq-VERT to detect divergent sequences
| Rotavirus gene | Sequence length (nt) | % mapped | Closest BLASTN hit identity (%) |
|---|---|---|---|
| VP1 | 3,280 | 97 | 78 |
| VP2 | 2,712 | 99 | 93 |
| VP3 | 2,592 | 86 | 78 |
| VP4 | 2,362 | 97 | 75 |
| NSP1 | 1,614 | 40 | 53 |
| VP6 | 1,194 | 92 | 96 |
| NSP3 | 1,075 | 95 | 76 |
| NSP2 | 954 | 88 | 96 |
| VP7 | 982 | 93 | 82 |
| NSP4 | 528 | 19 | 47 |
| NSP5 | 630 | 97 | 95 |
Detection of novel sequences.
To further test the capacity of VirCapSeq-VERT to detect novel viral sequences, we used an extract of a liver homogenate from a deer mouse experimentally infected with the rodent hepacivirus isolate RHVpl-01. The complete genome sequence of this isolate has a <65% global nucleotide sequence identity with the sequences used to design the VirCapSeq-VERT probes. Nonetheless, VirCapSeq-VERT selectively enriched RHVpl-01 sequence in conserved regions encoding the helicase and polymerase genes, for which bioinformatics analysis showed the presence of probes with up to 90% nucleotide identity in the VirCapSeq-VERT probe pool. We conclude therefore, from rotavirus and hepacivirus experiments, that while not an ideal platform for viral discovery, VirCapSeq-VERT can detect novel viruses through hybridization to short conserved sequence motifs within larger genome fragments.
Sample multiplexing.
During the estimation of the limit of detection of VirCapSeq-VERT, we processed samples individually or together during hybridization capture. The results obtained with the samples processed individually were superior (see Table S5 in the supplemental material). This finding suggested that competition for probe populations may compromise application in diagnostic settings where patient samples may have widely divergent virus loads. We investigated the practical impact of this potential confounding factor in assays using 21 barcoded libraries representing samples containing seven different viruses at genome loads that varied from 102 to 108. One set represented the seven different viruses each at a concentration of approximately 104 genome copies/library. To mimic competition anticipated in some clinical samples, the second set contained the same seven libraries at 104 copies, combined with an additional 14 libraries prepared with the seven viruses at 102 and at 105 to 108 copies. Virus detection was not impaired in multiplex assays even with samples that varied up to 104 in target concentration (Table 5); however, genome coverage was typically higher in 7-plex than in 21-plex assays.
TABLE 5 .
Genome mapping and coverage in VirCapSeq-VERT multiplex assays
| Virus | 7-plex mixa |
21-plex mix |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Load (copies) | %genome mapped | Avg coverage | Load (copies) | % genome mapped | Avg coverage | Load (copies) | % genome mapped | Avg coverage | Load (copies) | % genome mapped | Avg coverage | |
| HHV-1 | 104 | 100 | 4,258 | 104 | 99.6 | 583 | 106 | 99.9 | 5,438 | 102 | 84.5 | 10 |
| MERS-CoV | 104 | 27.9 | 1.1 | 104 | 20.1 | 0.34 | 106 | 98.7 | 23 | 102 | 0.3 | 0 |
| WNV | 104 | 98.8 | 4,785 | 104 | 98.9 | 251 | 108 | 100 | 7,799 | 102 | 99.1 | 107 |
| EBOV | 104 | 98.9 | 3,019 | 104 | 97.9 | 643 | 105 | 99.9 | 5,010 | 102 | 83.6 | 7 |
| EV-D68 | 104 | 99.9 | 6,644 | 104 | 99.8 | 4,816 | 106 | 99.9 | 6,911 | 102 | 91.8 | 64 |
| CVV-S | 104 | 100 | 6,197 | 104 | 100 | 2,364 | 107 | 100 | 7,332 | 102 | 99.8 | 49 |
| CVV-M | 104 | 100 | 7,603 | 104 | 100 | 1,048 | 107 | 100 | 7,798 | 102 | 100 | 23 |
| CVV-L | 104 | 100 | 2,409 | 104 | 100 | 242 | 107 | 100 | 7,735 | 102 | 93.4 | 4 |
| FLUAV -1 | 104 | 100 | 7,818 | 104 | 100 | 7,633 | 105 | 100 | 7,892 | 102 | 100 | 238 |
| FLUAV -2 | 104 | 100 | 7,904 | 104 | 100 | 7,741 | 105 | 100 | 7,902 | 102 | 100 | 575 |
| FLUAV -3 | 104 | 100 | 7,792 | 104 | 100 | 7,658 | 105 | 100 | 7,906 | 102 | 100 | 276 |
| FLUAV -4 | 104 | 100 | 7,800 | 104 | 100 | 7,584 | 105 | 100 | 7,799 | 102 | 100 | 594 |
| FLUAV -5 | 104 | 100 | 7,747 | 104 | 100 | 7,605 | 105 | 100 | 7,746 | 102 | 100 | 352 |
| FLUAV -6 | 104 | 100 | 7,721 | 104 | 100 | 7,560 | 105 | 100 | 7,721 | 102 | 100 | 358 |
| FLUAV -7 | 104 | 100 | 7,355 | 104 | 100 | 7,100 | 105 | 100 | 7,711 | 102 | 100 | 251 |
| FLUAV -8 | 104 | 100 | 7,367 | 104 | 100 | 7,360 | 105 | 100 | 7,367 | 102 | 100 | 397 |
qPCR quantitated nucleic acid extracts representing seven different viruses were used to spike a background of human blood nucleic acid at levels of approximately 104 copies/100 ng, 102 copies/100 ng, and 105 to 108 copies/100 ng. Individual sequence libraries were prepared using 21 different indexes for bar coding. Libraries were mixed for capture hybridization into a 7-plex mix (libraries prepared from 104 loads) and the complete 21-plex mix.
To determine the utility of VirCapSeq-VERT in characterization of virome diversity and dynamics, we analyzed a set of 23 serum samples collected from multiply transfused hemophilia patients known to contain hepatitis C virus (HCV), GB virus C (GBV-C), human immunodeficiency virus (HIV), and torque teno virus (TTV). Samples were amplified using unique bar codes, and two pools were generated for VirCapSeq-VERT. Pool 1 contained 9 samples. Pool 2 contained the same 9 samples mixed with the remaining 14 samples. All viruses in the 9-plex as well as in the 23-plex sample pool were efficiently characterized (see Fig. S1 in the supplemental material).
DISCUSSION
Sequencing approaches have enabled metagenomics, including virome studies, and are of increasing interest in the field of diagnostics. However, concerns regarding sensitivity, especially in high-host-background settings, cumbersome and time-consuming sample processing, and cost pose hurdles that would need to be overcome in order to realize the potential of HTS. Unlike 16S rRNA of bacteria, viruses lack universally conserved markers and have plastic genomes that easily generate mutants, strains, and variants. Virus variants differing in sequence, even by a single point mutation, can vary in host range, transmissibility and pathogenicity (23, 24). Accordingly, an ideal viral diagnostic platform should enable sensitive multiplexed detection of all viruses and their variants. Nucleic acid capture with oligonucleotides has been used to enhance the efficiency of HTS for characterizing host (25, 26) or selected microbial (27, 28) targets at low scale. However, to our knowledge, VirCapSeq-VERT is the first example wherein a positive selection method for HTS has been employed for comprehensive, sensitive application in microbial diagnostics and whole virome analysis.
Current virus diagnostic assays that are commonly based on PCR assays targeting one or a few specific agents may fail to detect virus variants and provide only limited genotypic information. VirCapSeq-VERT addresses many of the current challenges of PCR and HTS for diagnostics and virome analysis. The sensitivity and specificity of the VirCapSeq-VERT are comparable to those of agent-specific real-time PCR (see Table S6 in the supplemental material). Additionally, the 100- to 10,000-fold increase achieved in on-target reads enables leveraging of sequencing depth against costs in research applications. The approximately per-sample cost of 40 U.S. dollars (USD) of VirCapSeq-VERT in a 20-plex sample format compares favorably with costs of other enrichment procedures, such as rRNA depletion (approximately 65 USD per sample), particularly given its advantages in sensitivity, genome coverage, and ease of use. The capacity for highly multiplexed sample processing and simplified sample handling is cost-effective and reduces the risk of cross contamination.
The VirCapSeq-VERT system is not specifically designed for viral discovery; nonetheless, it enables sequencing of genomes with as little as 75% overall sequence identity. Results of our rotavirus and hepacivirus analyses indicate that where the goal is detection rather than comprehensive genome sequencing, VirCapSeq-VERT has the potential, through hybridization to conserved regions, to detect novel viruses with an overall nucleotide divergence in the range of 40%.
In summary, VirCapSeq-VERT has promise as a tool for diagnostic and research applications. It has sensitivity similar to that obtained with targeted real-time PCR, with the advantage of detecting viral variants that would not be captured with specific PCR assays as well as the potential to provide the complete genome sequence needed for assessment of viral diversity and evolution for epidemiological and public health applications.
MATERIALS AND METHODS
Samples and specimens.
Facsimiles of clinical specimens were generated in a background of NA extracted from normal human lung tissue, EDTA-blood, or serum. The samples were spiked with viral NA and quantitated by virus-specific TaqMan real-time (reverse transcription) PCR (qPCR). NA from cell culture or blood, serum, or tissue samples was extracted using the easyMAG system (bioMérieux, Marcy l’Etoile, France) or AllPrep DNA/RNA kits (Qiagen, Hilden, Germany). Background NA was quantitated by NanoDrop (Wilmington, DE, USA) or Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA) and mixed with variable quantities of viral NA derived from enterovirus D68 (EV-D68) (29), West Nile virus (WNV) (39), dengue virus 3 (DENV-3) (40), and Middle East respiratory syndrome coronavirus (MERS-CoV) (41), representing single-strand, positive-sense RNA viruses of different genome sizes; Ebola virus (EBOV) influenza A virus H3N2 (FLUAV, A/Moscow/10/99; WHO Influenza Centre, MRC, London, United Kingdom) and Cache Valley virus (CVV) (42), representing nonsegmented and segmented negative-strand RNA viruses; and herpes simplex virus 1 (HHV-1, ATCC VR-733), as a large double-strand DNA virus. Spiking was performed using NA stocks banked at the Center of Infection and Immunity originally derived from virus cultures or positive diagnostic specimens, with the exception of EBOV, which was provided as noninfectious nucleic acid extract by Peter Jahrling at the National Institute of Allergy and Infectious Diseases, National Institutes of Health. TaqMan PCR primer and probes for the various viruses are cited or available on request.
To determine the limit of detection and to assess VirCapSeq-VERT in comparison to conventional target enrichment procedures, normal human lung tissue homogenate, EDTA-blood, or serum samples were spiked with different amounts of live EV-D68, HHV-1, and FLUAV stock quantitated by qPCR.
Clinical samples included a human nasal swab sample known to be positive for EV-D68 (30), liver specimens from deer mice infected with deer mouse hepacivirus (31), a sample of bat feces pellets in which rotaviral sequences had been identified (unpublished), and serum samples from hemophilia patients coinfected with hepatitis C virus (HCV), GB virus C (GBV-C), human immunodeficiency virus (HIV), and torque teno virus (TTV).
Selection of probe sequences.
The EMBL Coding Domain Sequence database (release 122, December 2014; ftp://ftp.ebi.ac.uk/pub/databases/embl/cds/release/std/), containing 2,199,467 records was clustered at 96% sequence identity by CD-Hit (32), yielding a database of 401,716 representative sequences spanning all virus sequence records, excluding bacteriophages. A list of all virus genera known to infect vertebrates was generated from the master species list of the International Committee on Taxonomy of Viruses (ICTV; http://talk.ictvonline.org/files/ictv_documents/m/msl/5208.aspx). Through cross-referencing of protein IDs with NCBI taxonomy IDs, a set of 342,438 coding sequence records was identified for the selected virus genera. The sequences were broken into fragments, clustered at 90% sequence identity, and used to generate 100-nt probe sequences that were tiled across the genes at approximately 25- to 50-nt intervals. A library of 1,993,200 oligonucleotide probes was selected. The NimbleGen cleavable array platform was employed for synthesis of the biotinylated, soluble probe library (SeqCap EZ Choice; Roche/NimbleGen, Basel, Switzerland), and probe sequences were refined by adjusting their lengths to conform to NimbleGen synthesis parameters, such as maximum Tm or homopolymer repeat length.
Conventional target sequence enrichment procedures.
Conventional virus enrichment methods commonly used in metagenomic sequencing-based virus discovery include filtration and pre-extraction nuclease treatments, often combined with postextraction DNase I and/or depletion of ribosomal rRNA sequences. Briefly, samples (100 to 300 µl) were filtered through 0.45-μm-pore-size sterile disk filters (Merck/Millipore, Billerica, MA, USA) to enrich for viruses over cells or bacteria. The flowthrough was treated with 1 μl RNase A (10 mg/ml; Thermo, Fisher, Waltham, MA, USA) for 15 min at room temperature, followed by a cocktail of 8 U Turbo DNase (Thermo, Fisher), 250 U Benzonase (Merck/Millipore), and 10 mM MgCl2 for 45 min at room temperature to digest non-particle-protected NAs. Protected NAs, such as those in viral particles, were extracted by easyMAG (bioMérieux) or AllPrep kits (Qiagen). Postextraction digestion by DNase I (2 U/µg DNA for 15 min at 37oC; Thermo, Fisher) was added in some instances to digest chromosomal DNA (cellular and bacterial), but it will also digest viral DNA (e.g., HHV-1 DNA), whereas mRNA transcripts generated from actively replicating cellular virus would be maintained. Depletion of nondesired host mRNA sequences was achieved using RiboZero magnetic kits (Illumina, San Diego, CA, USA). Enriched preparations were subjected to reverse transcription and sequence library preparation.
Conventional HTS.
Total NA extracts were reverse transcribed using SuperScript III (Thermo, Fisher) with random hexamers. The cDNA was RNase H treated prior to second-strand synthesis with Klenow fragment (New England Biolabs, Ipswich, MA, USA). The generated double-stranded cDNA was sheared to an average fragment size of 200 bp using the manufacturer’s standard settings (E210 focused ultrasonicator; Covaris, Woburn, MA, USA). Sheared product was purified (AxyPrep Mag PCR cleanup beads; Axygen/Corning, Corning, NY, USA), and libraries were constructed using KAPA library preparation kits (KAPA, Wilmington, MA, USA). For NA input quantities of 10 to 100 ng double-stranded cDNA, the cycle number of the final PCR amplification was increased to 12 cycles, instead of 9 cycles for samples with >100 ng double-stranded cDNA. Final products were purified (AxyPrep) and quantitated by Bioanalyzer (Agilent) for Illumina sequencing.
Virome capture sequencing.
Libraries were prepared by essentially following the standard KAPA protocol but including viral sequence capture, following mainly the SeqCap RNA enrichment system protocol (Roche/NimbleGen). Briefly, total NA extract was reverse transcribed using SuperScript III (Thermo, Fisher) with random hexamers. The cDNA was RNase H treated prior to second-strand synthesis with Klenow fragment (New England Biolabs). The resulting double-stranded cDNA/DNA mix was sheared to an average fragment size of 200 bp using the manufacturer’s standard settings (Covaris E210 focused ultrasonicator). Sheared product was purified (AxyPrep), and libraries were constructed using KAPA library preparation kits (KAPA) with Roche/NimbleGen adapter kits. The quality and quantity of libraries were checked using a Bioanalyzer (Agilent). The libraries were then mixed with a SeqCap HE universal oligonucleotide, SeqCap HE index oligonucleotides, and COT DNA and vacuum evaporated at 60°C for approximately 40 min. Dried samples were mixed with 2× hybridization buffer and hybridization component A (Roche/NimbleGen) prior to denaturation at 95°C for 10 min. The VirCap probe library (4.5 μl) was added and hybridized at 47°C for 12 h in a standard PCR thermocycler. SeqCap Pure capture beads (Roche/NimbleGen) were washed twice, mixed with the hybridization mix, and kept at 47°C for 45 min with vortexing for 10 s every 10 to 15 min. The streptavidin capture beads complexed with biotinylated VirCapSeq-VERT probes were trapped (DynaMag-2 magnet; Thermo, Fisher) and washed once at 47°C and then twice more at room temperature with wash buffers of increasing stringency. Finally, beads were suspended in 50 µl water and directly subjected to posthybridization PCR (SeqCap EZ accessory kit V2; Roche/NimbleGen). The PCR products were purified (Agencourt Ampure DNA purification beads; Beckman Coulter, Brea, CA, USA) and quantitated by Bioanalyzer (Agilent) for Illumina sequencing.
Data analysis and bioinformatics pipeline.
Sequencing on the Illumina HiSeq 2500 platform (Illumina) resulted in an average of 210 million reads per lane. Samples were demultiplexed using Illumina software, and FastQ files were generated. Demultiplexed and Q30-filtered FastQ files were mapped against reference genomes from GenBank with Bowtie2 mapper 2.0.6 (http://bowtie-bio.sourceforge.net). SAMtools (v 0.1.19) (33) were used to generate the consensus genomes and coverage statistics. Integrative Genomics Viewer (v 2/3/55) (34) was used to generate coverage plots. Host background levels were determined from Bowtie2 mappings against the host genomes downloaded from the NCBI. Sequencing data obtained from the unknown samples was preprocessed using PRINSEQ (v 0.20.2) (35) software, and filtered reads were aligned against the host reference databases to remove the host background. The resulting reads were de novo assembled using MIRA (v 4.0) (36) or SOAPdenovo2 (v 2.04) (37) assemblers, and contigs and unique singletons were subjected to homology search using MegaBlast against the GenBank nucleotide database; sequences that showed poor or no homology at the nucleotide level were screened by BLASTX against the viral GenBank protein database. Viral sequences from BLASTX analysis were subjected to another round of BLASTX homology search against the entire GenBank protein database to correct for biased E values and taxonomic misassignments. Based on the contigs identified for different viral strains, GenBank sequences were downloaded and used for mapping the whole data set to recover partial or complete genomes. Viral read numbers were obtained from counting the number of reads mapping to contig sequences and unassembled singletons, and percentages were calculated in relation to the total read number obtained. Percentages were converted into heatmaps using MultiExperiment Viewer (MeV v4.9) (38).
SUPPLEMENTAL MATERIAL
Study of virome composition and dynamics by VirCapSeq-VERT. Download
Virus taxa selected for VirCapSeq-VERT probe design.
VirCapSeq-VERT probe parameters.
Probe coverage for selected taxa.
VirCapSeq-VERT increases the efficiency of high-throughput sequencing for virus detection and characterization.
Estimation of the limit of detection achieved by VirCapSeq-VERT using nucleic acid extracts.
Estimation of the limit of detection achieved by VirCapSeq-VERT using live enterovirus D68 (EV-D68).
Improvement of genome sequence recovery and coverage with VirCapSeq-VERT.
Efficiency of enterovirus D68 (EV-D68) detection and genome sequencing in nasal swab using VirCapSeq-VERT or other methods for viral template enrichment.
Efficiency of rotavirus detection and genome sequencing in bat feces using VirCapSeq-VERT or other methods for viral template enrichment.
ACKNOWLEDGMENTS
We thank Alexandra Petrosov for expert sequence library preparation, Parisa Zolfaghari for virus culture and generation of PCR standards, James Ng for executing PCR assays, Lokendra Singh Chauhan for sample preparation, Rafal Tokarz for EV-D68 qPCR, and Peter Jahrling for Ebola virus nucleic acid extract. We thank Bohyun Lee and Adrian Caciula for excellent bioinformatics support and Ellie Kahn for assistance with the manuscript.
The study was supported by the National Institutes of Health (U19 AI109761 to W.I.L., T.B., A.K., N.M., K.J., and A.K. and R01 HL119485 to A. Kapoor) and the Department of Homeland Security (KSUNI S11045.01 to T.B.).
Footnotes
Citation Briese T, Kapoor A, Mishra N, Jain K, Kumar A, Jabado OJ, Lipkin WI. 2015. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. mBio 6(5):e01491-15. doi:10.1128/mBio.01491-15.
REFERENCES
- 1.Kapoor A, Lipkin WI. 2001. Virus discovery in the 21st century. In eLS. John Wiley & Sons, New York, NY. [Google Scholar]
- 2.Mullis KB, Faloona FA. 1987. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol 155:335–350. [DOI] [PubMed] [Google Scholar]
- 3.VanDevanter DR, Warrener P, Bennett L, Schultz ER, Coulter S, Garber RL, Rose TM. 1996. Detection and analysis of diverse herpesviral species by consensus primer PCR. J Clin Microbiol 34:1666–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casas I, Tenorio A, Echevarría JM, Klapper PE, Cleator GM. 1997. Detection of enteroviral RNA and specific DNA of herpesviruses by multiplex genome amplification. J Virol Methods 66:39–50. doi: 10.1016/S0166-0934(97)00035-9. [DOI] [PubMed] [Google Scholar]
- 5.Liljas L, Tate J, Lin T, Christian P, Johnson JE. 2002. Evolutionary and taxonomic implications of conserved structural motifs between picornaviruses and insect picorna-like viruses. Arch Virol 147:59–84. doi: 10.1007/s705-002-8303-1. [DOI] [PubMed] [Google Scholar]
- 6.Briese T, Palacios G, Kokoris M, Jabado O, Liu Z, Renwick N, Kapoor V, Casas I, Pozo F, Limberger R, Perez-Brena P, Ju J, Lipkin WI. 2005. Diagnostic system for rapid and sensitive differential detection of pathogens. Emerg Infect Dis 11:310–313. doi: 10.3201/eid1102.040492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sampath R, Hofstadler SA, Blyn LB, Eshoo MW, Hall TA, Massire C, Levene HM, Hannis JC, Harrell PM, Neuman B, Buchmeier MJ, Jiang Y, Ranken R, Drader JJ, Samant V, Griffey RH, McNeil JA, Crooke ST, Ecker DJ. 2005. Rapid identification of emerging pathogens: coronavirus. Emerg Infect Dis 11:373–379. doi: 10.3201/eid1103.040629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunstein J, Thomas E. 2006. Direct screening of clinical specimens for multiple respiratory pathogens using the Genaco respiratory panels 1 and 2. Diagn Mol Pathol 15:169–173. doi: 10.1097/01.pdm.0000210430.35340.53. [DOI] [PubMed] [Google Scholar]
- 9.Ecker DJ, Sampath R, Massire C, Blyn LB, Hall TA, Eshoo MW, Hofstadler SA. 2008. Ibis T5000: a universal biosensor approach for microbiology. Nat Rev Microbiol 6:553–558. doi: 10.1038/nrmicro1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gadsby NJ, Hardie A, Claas EC, Templeton KE. 2010. Comparison of the Luminex respiratory virus panel fast assay with in-house real-time PCR for respiratory viral infection diagnosis. J Clin Microbiol 48:2213–2216. doi: 10.1128/JCM.02446-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang P, Pardee AB. 1992. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science 257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 12.Lisitsyn N, Lisitsyn N, Wigler M. 1993. Cloning the differences between two complex genomes. Science 259:946–951. doi: 10.1126/science.8438152. [DOI] [PubMed] [Google Scholar]
- 13.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 14.Lipkin WI, Travis GH, Carbone KM, Wilson MC. 1990. Isolation and characterization of Borna disease agent cDNA clones. Proc Natl Acad Sci U S A 87:4184–4188. doi: 10.1073/pnas.87.11.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Briese T, Jia XY, Huang C, Grady LJ, Lipkin WI. 1999. Identification of a Kunjin/West Nile-like flavivirus in brains of patients with New York encephalitis. Lancet 354:1261–1262. doi: 10.1016/S0140-6736(99)04576-6. [DOI] [PubMed] [Google Scholar]
- 16.Allander T, Emerson SU, Engle RE, Purcell RH, Bukh J. 2001. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proc Natl Acad Sci U S A 98:11609–11614. doi: 10.1073/pnas.211424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Victoria JG, Kapoor A, Dupuis K, Schnurr DP, Delwart EL. 2008. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathog 4:e1000163. doi: 10.1371/journal.ppat.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kapoor A, Victoria J, Simmonds P, Wang C, Shafer RW, Nims R, Nielsen O, Delwart E. 2008. A highly divergent picornavirus in a marine mammal. J Virol 82:311–320. doi: 10.1128/JVI.01240-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 20.Uchida T, Suzuki K, Hayashi N, Iida F, Hara T, Oo SS, Wang CK, Shikata T, Ichikawa M, Rikihisa T. 1992. Hepatitis E virus: cDNA cloning and expression. Microbiol Immunol 36:67–79. doi: 10.1111/j.1348-0421.1992.tb01643.x. [DOI] [PubMed] [Google Scholar]
- 21.Wang D, Coscoy L, Zylberberg M, Avila PC, Boushey HA, Ganem D, DeRisi JL. 2002. Microarray-based detection and genotyping of viral pathogens. Proc Natl Acad Sci U S A 99:15687–15692. doi: 10.1073/pnas.242579699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palacios G, Quan PL, Jabado OJ, Conlan S, Hirschberg DL, Liu Y, Zhai J, Renwick N, Hui J, Hegyi H, Grolla A, Strong JE, Towner JS, Geisbert TW, Jahrling PB, Büchen-Osmond C, Ellerbrok H, Sanchez-Seco MP, Lussier Y, Formenty P, Nichol MS, Feldmann H, Briese T, Lipkin WI. 2007. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerg Infect Dis 13:73–81. doi: 10.3201/eid1301.060837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guillot S, Otelea D, Delpeyroux F, Crainic R. 1994. Point mutations involved in the attenuation/neurovirulence alternation in type 1 and 2 oral polio vaccine strains detected by site-specific polymerase chain reaction. Vaccine 12:503–507. doi: 10.1016/0264-410X(94)90307-7. [DOI] [PubMed] [Google Scholar]
- 24.Drexler JF, Grard G, Lukashev AN, Kozlovskaya LI, Böttcher S, Uslu G, Reimerink J, Gmyl AP, Taty-Taty R, Lekana-Douki SE, Nkoghe D, Eis-Hübinger AM, Diedrich S, Koopmans M, Leroy EM, Drosten C. 2014. Robustness against serum neutralization of a poliovirus type 1 from a lethal epidemic of poliomyelitis in the Republic of Congo in 2010. Proc Natl Acad Sci U S A 111:12889–12894. doi: 10.1073/pnas.1323502111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mercer TR, Clark MB, Crawford J, Brunck ME, Gerhardt DJ, Taft RJ, Nielsen LK, Dinger ME, Mattick JS. 2014. Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nat Protoc 9:989–1009. doi: 10.1038/nprot.2014.058. [DOI] [PubMed] [Google Scholar]
- 26.Clark MB, Mercer TR, Bussotti G, Leonardi T, Haynes KR, Crawford J, Brunck ME, Cao KA, Thomas GP, Chen WY, Taft RJ, Nielsen LK, Enright AJ, Mattick JS, Dinger ME. 2015. Quantitative gene profiling of long noncoding RNAs with targeted RNA sequencing. Nat Methods 12:339–342. doi: 10.1038/nmeth.3321. [DOI] [PubMed] [Google Scholar]
- 27.Bent ZW, Tran-Gyamfi MB, Langevin SA, Brazel DM, Hamblin RY, Branda SS, Patel KD, Lane TW, VanderNoot VA. 2013. Enriching pathogen transcripts from infected samples: a capture-based approach to enhanced host-pathogen RNA sequencing. Anal Biochem 438:90–96. doi: 10.1016/j.ab.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Depledge DP, Palser AL, Watson SJ, Lai IY, Gray ER, Grant P, Kanda RK, Leproust E, Kellam P, Breuer J. 2011. Specific capture and whole-genome sequencing of viruses from clinical samples. PLoS One 6:e27805. doi: 10.1371/journal.pone.0027805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown BA, Nix WA, Sheth M, Frace M, Oberste MS. 2014. Seven strains of enterovirus D68 detected in the United States during the 2014 severe respiratory disease outbreak. Genome Announc 2:e01201-14. doi: 10.1128/genomeA.01201-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tokarz R, Firth C, Madhi SA, Howie SR, Wu W, Sall AA, Haq S, Briese T, Lipkin WI. 2012. Worldwide emergence of multiple clades of enterovirus 68. J Gen Virol 93:1952–1958. doi: 10.1099/vir.0.043935-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kapoor A, Simmonds P, Scheel TK, Hjelle B, Cullen JM, Burbelo PD, Chauhan LV, Duraisamy R, Sanchez-Leon M, Jain K, Vandegrift KJ, Calisher CH, Rice CM, Lipkin WI. 2013. Identification of rodent homologs of hepatitis C virus and pegiviruses. mBio 4:e00216-13. doi: 10.1128/mBio.00216-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 33.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. BioInformatics 27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chevreux B, Wetter T, Suhai S. 1999. Genome sequence assembly using trace signals and additional sequence information. Comput Sci Biol 99:45–56. [Google Scholar]
- 37.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J, Wu G, Zhang H, Shi Y, Liu Y, Yu C, Wang B, Lu Y, Han C, Cheung DW, Yiu SM, Peng S, Xiaoqian Z, Liu G, Liao X, Li Y, Yang H, Wang J, Lam TW, Wang J. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. 2003. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378. [DOI] [PubMed] [Google Scholar]
- 39.Briese T, Glass WG, Lipkin WI. 2000. Detection of West Nile virus sequences in cerebrospinal fluid. Lancet 355:1614–1615. doi: 10.1016/S0140-6736(00)02220-0. [DOI] [PubMed] [Google Scholar]
- 40.Nunes MR, Palacios G, Faria NR, Sousa EC Jr., Pantoja JA, Rodrigues SG, Carvalho VL, Medeiros DB, Savji N, Baele G, Suchard MA, Lemey P, Vasconcelos PF, Lipkin WI. 2014. Air travel is associated with intracontinental spread of dengue virus serotypes 1–3 in Brazil. PLoS Negl Trop Dis 8:e2769. doi: 10.1371/journal.pntd.0002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Briese T, Mishra N, Jain K, Zalmout IS, Jabado OJ, Karesh WB, Daszak P, Mohammed OB, Alagaili AN, Lipkin WI. 2014. Middle East respiratory syndrome coronavirus quasispecies that include homologues of human isolates revealed through whole-genome analysis and virus cultured from dromedary camels in Saudi Arabia. mBio 5:e01146-14. doi: 10.1128/mBio.01146-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Briese T, Kapoor V, Lipkin WI. 2007. Natural M-segment reassortment in Potosi and Main drain viruses: implications for the evolution of orthobunyaviruses. Arch Virol 152:2237–2247. doi: 10.1007/s00705-007-1069-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Study of virome composition and dynamics by VirCapSeq-VERT. Download
Virus taxa selected for VirCapSeq-VERT probe design.
VirCapSeq-VERT probe parameters.
Probe coverage for selected taxa.
VirCapSeq-VERT increases the efficiency of high-throughput sequencing for virus detection and characterization.
Estimation of the limit of detection achieved by VirCapSeq-VERT using nucleic acid extracts.
Estimation of the limit of detection achieved by VirCapSeq-VERT using live enterovirus D68 (EV-D68).
Improvement of genome sequence recovery and coverage with VirCapSeq-VERT.
Efficiency of enterovirus D68 (EV-D68) detection and genome sequencing in nasal swab using VirCapSeq-VERT or other methods for viral template enrichment.
Efficiency of rotavirus detection and genome sequencing in bat feces using VirCapSeq-VERT or other methods for viral template enrichment.