

Abstract
Iron is an essential element involved in various biological pathways. When present in excess within the cell, iron can be toxic due to its ability to catalyse the formation of damaging radicals, which promote cellular injury and cell death. Within the liver, iron related oxidative stress can lead to fibrosis and ultimately to cirrhosis. Here we review the role of excessive iron in the pathologies associated with various chronic diseases of the liver. We also describe the molecular mechanism by which iron contributes to the development of hepatic fibrosis.
Keywords: Iron, Fibrosis, Oxidative stress, Hepatic stellate cell, Haemochromatosis, Hepatitis C, Non-alcoholic fatty liver disease, Alcoholic liver disease
INTRODUCTION
Iron, created by stellar nucleosynthesis, is the most abundant element on Earth in terms of mass, making up 35% of total planetary mass. Iron, in its ferrous (Fe2+) and ferric (Fe3+) forms, is critical to all life forms from the simplest filamentous algae through to the most complex multicellular organisms. Iron is an essential element mainly present within the cells in association with haemoprotein (haemoglobin, myoglobin) or within an iron-sulphur cluster of various metalloproteins (e.g., aconitase and Rieske proteins of the respiratory chain)[1]. Iron is involved in the redox-driven processes of oxygen transport, electron transport and various enzymatic reactions such as DNA synthesis, transcriptional regulation, catalysis as well as nitric oxide (NO) and oxygen sensing[1]. Mammals do not have any major physiological pathway for iron excretion. Therefore, iron uptake and storage is closely regulated so as to avoid deficiency, and excess. In humans, iron deficiency is manifested as anaemia and with increasing severity can eventually result in cardiac failure. Iron overload, especially at sites of storage, enhances oxidative stress ultimately leading to lipid, nucleic acid and protein peroxidation. Within the liver, which is the major site of iron storage, enhanced oxidative stress can lead to fibrosis, cirrhosis, hepatocellular carcinoma (HCC) and death.
In this review, we outline the hepatic disease states where iron is an important factor in disease progression. We also discuss the role of iron in promoting liver fibrosis and those cells and mechanisms most important in the underlying wound healing/fibrotic processes.
HEPATIC FIBROSIS
Hepatic fibrosis is promoted by various pathogenic, mechanical and toxic insults to the liver, and is part of a physiological wound healing response. If the injurious stimuli are chronic, the degree of fibrosis worsens, leading to cirrhosis, and eventually to hepatic failure and death. The interactions between various resident hepatic cell populations and immune cells that lead to the establishment of fibrosis are complex, and not yet fully understood. However, some profibrogenic pathways and clinical outcomes are common to several disease states, and these will be briefly outlined below.
In the normal liver, a balance is struck between extracellular matrix (ECM) deposition and degradation, a process that is tightly regulated by matrix metalloproteinases (MMP) and their specific inhibitors (TIMPs)[2]. Fibrosis is associated with major quantitative and qualitative changes in the ECM. These changes are mainly due to an increase in the expression of TIMP-1[3] and an increase in the expression of various ECM components, which include fibrillary collagensIand III, collagen IV, fibronectin, elastin and laminin[4].
The hepatic stellate cell (HSC) resides within the space of Disse, and is responsible for the majority of ECM deposition in the normal and fibrotic liver[5,6]. In the normal liver the primary role of the HSC is the regulation of vitamin A homeostasis and storage[6]. In times of chronic injury, the HSC transdifferentiates into a myofibroblastic cell exhibiting contractile, proliferative, inflammatory and fibrogenic properties. Transdifferentiation occurs in response to soluble fibrogenic and proliferative factors released mainly by Kupffer cells (KC), (PDGF, TGFβ1) or by damaged hepatocytes (IGF-1, TNFα, EGF)[2]. Once transdifferentiated, the HSC expresses a number of myogenic markers (including α smooth muscle actin (αSMA), c-myb and myocyte enhancer factor-2) that allows them to be readily identified by immunohistochemical techniques[4]. In addition, HSC also express a number of neuroendocrine proteins (e.g., glial fibrillary acidic protein or GFAP, synaptophysin and nestin[4]) and receptors for various neurotransmitters[7]. Once activated, HSC also release TGFβ1 and PDGF-BB, thereby ensuring a self-sustained phenotypic change[8,9]. Fibrosis was once thought to be irreversible; however, a growing body of evidence now suggests that once the underlying source of liver injury is treated, fibrosis, and even cirrhosis, to some extent, are reversible. Fibrosis resolution is known to be accompanied by HSC apoptosis, and to some degree of reversion of HSC to their original phenotype[10].
IRON AND DISEASES OF THE LIVER
Iron is proposed to play a role in promoting hepatic fibrosis in a number of different chronic liver disease states. The evidence supporting or contradicting those propositions is outlined in the following section.
Haemochromatosis
Haemochromatosis is a term used to describe excessive iron loading of the liver leading ultimately to cirrhosis and frequently to HCC[11]. The knowledge regarding the underlying causes of haemochromatosis is gradually expanding, and the term has now been subdivided according to the specific genetic mutation involved. Iron loading of the liver is a life long process in patients with haemochromatosis, occurring without any apparent overt inflammation or elevation of serum liver enzymes such as alanine aminotransferase (ALT) and aspartate aminotransferase (AST). The symptoms associated with hereditary haemochromatosis (HH) include; hepatomegaly, malaise, insulin resistance and arthritis, to name but a few. When hepatic iron reaches the threshold concentration of 60 μmol/g dry weight, HSC begin to exhibit the early stages of cellular activation (namely αSMA expression), a key event in the initiation of hepatic fibrosis[12]. Evidence also suggests that there is a hepatic iron concentration threshold (about 250 μmol/g dry weight) beyond which cirrhosis can develop in patients with HH[13,14]. Initial iron loading of the liver occurs in hepatocytes located in Rappaport zone 1, progressively extending to hepatocytes in zones 2 and 3. In the later stages of the disease, KC also accumulate iron and it is the co-loading of both hepatocytes and KC that is, believed to allow fibrosis to become established[15]. Patients with well established hepatic iron loading are also at a significantly greater risk of developing HCC[11]. Phlebotomy is widely used to reduce iron burden in patients with haemochromatosis and, is also an effective way to reverse hepatic fibrosis[14,16], although regression of cirrhosis remains controversial[14,16].
Transition of the iron-loaded liver from non-fibrotic to fibrotic, and then onwards to cirrhotic is not always a clear-cut process, and there is growing evidence to suggest that other factors, such as excessive alcohol consumption[17], viral hepatitis[18] and steatosis[19] among others, have an important role in that transition. In fact, iron overload has been shown to be important in the pathology of other liver disease such as non-alcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD) and chronic hepatitis C.
Non-alcoholic fatty liver disease
Increase in caloric intake and an evermore sedentary life-style have led to a surge in the prevalence of people being overweight or obese. Current estimates suggest that as much as 60% of the over 18 age group in Australia will be overweight or obese by the year 2010[20]. Obesity impacts the liver in the form of NAFLD, which is now recognised as the most common form of liver disease. NAFLD begins with steatosis (fat accumulation within the liver) and can remain in this state without any apparent associated pathology. In its more advanced form NAFLD is often referred to as non-alcoholic steatohepatitis (NASH). It is believed that in order for steatosis to advance to NASH a secondary liver insult is also required often termed the “second hit”[21]. Iron is among several co-factors known to increase hepatic oxidative stress, and constitutes a “second hit” agent that is postulated to contribute to the progression of steatosis to NASH.
Several studies have highlighted mutations (C282Y and H63D) in the HFE gene that positively correlate with the presence of NAFLD/NASH[19,22-24]. These studies suggest that while combined NASH/NAFLD and HFE mutation have a negative effect on disease severity as highlighted by serum ALT concentrations and fibrosis grade, hepatic iron concentration is not always elevated. Interestingly Mendler and colleagues also noted that patients with an elevated iron burden almost always displayed insulin resistance irrespective of liver damage[24]. Insulin resistance is known to play a central role in the accumulation of triglycerides within the hepatocytes, and in the initiation of the inflammatory cascade ultimately leading to cirrhosis[25]. The correlation between elevated hepatic iron and disease severity in patients with NAFLD/NASH is not always consistent. Several studies have failed to find any evidence of increased hepatic iron levels in patients with NAFLD/NASH, with histological severity being linked to the presence of ballooning hepatocyte degeneration, Mallory hyaline, older age, obesity and the presence of diabetes mellitus[26-28]. The role of iron in the pathologies associated with NAFLD is, however, further supported by several studies that demonstrate the beneficial effects of iron depletion on serum ALT concentrations and insulin response in patients with NAFLD[29]. How and why steatosis and iron combined lead to exacerbated fibrosis is possibly linked to the oxidative stress that both can exert.
Alcoholic liver disease
ALD is caused by high risk alcohol consumption over a number of years. Of those engaging in high risk alcohol consumption, only approximately 30% go on to develop liver cirrhosis[21]. This would suggest that other factors influence disease severity and progression in patients with ALD. Iron is one such factor known to affect the pathogenesis of ALD. In pre-cirrhotic ALD, approximately 29% of patients demonstrate an elevated hepatic iron concentration[30]. As the disease progresses to cirrhosis, as many as 57% of patients have iron overload (> 25 μmol/g)[31]. It is also apparent that iron is an independent risk factor for the development of fibrosis[32] in ALD, and higher iron concentrations also correlate with reduced survival[33]. Alcohol has been postulated to enhance iron uptake by a number of different mechanisms, which have recently been reviewed by Brittenham[34]. In addition, recent work by Bridle and colleagues suggest that ethanol may perturb IL-6-mediated hepcidin expression resulting in enhanced absorption of iron and hepatosiderosis[35]. This is important when one considers that mammals lack an effective mechanism by which excess iron can be eliminated from the body[34].
Hepatitis C
Viral hepatitis encompasses a range of different entities from hepatitis A through E, which are caused by distinct viruses. Of these, hepatitis B and C are perhaps the most significant in terms on patient morbidity and mortality. Hepatitis B virus (HBV) is a member of the Hepadnaviridae family. Estimates suggest that between 3% and 6% of the world’s population are infected with HBV, with up to one third having been previously exposed[36]. Hepatitis C virus (HCV) is a member of the Flaviviridae family and is thought to infect somewhere between 150 and 200 million people worldwide. Both types of viral hepatitis contribute to the development of cirrhosis and HCC. As is common with many forms of liver disease, the severity and rate at which fibrosis progresses are influenced by a great number of external factors and in viral hepatitis these include male gender, alcohol consumption, iron status and age at infection[37].
The association between elevated serum iron and hepatitis B was first described by Blumberg et al in 1981[38]. Some years later, a link was also found between serum iron, ferritin, transferrin saturation levels and HCV infection[39,40]. However, these results did not correlate with an increased hepatic iron concentration. Where hepatic iron was found to be elevated in association with HCV infection (in 5% of cases), it was seldom to levels deemed hepatotoxic[39]. In HCV-infected patients, iron is deposited in hepatocytes, sinusoidal cells, and portal mesenchymal cells, with the degree of portal mesenchymal iron deposition correlating with both hepatic inflammation and fibrosis[41]. Martinelli and colleagues demonstrated a significant correlation between liver iron scores and the number of GFAP and αSMA-positive HSC in patients with HCV. These αSMA-positive or “activated HSC” were located primarily in Rappaport zones 1 and 3[42]. Similar findings were also reported by Rigamonti and colleagues, who also described a correlation between hepatic iron concentration and HSC activation. They suggested that, in patients with HCV infection, iron was important in both HSC activation and fibrosis progression[43]. Besides being directly correlated with inflammation and fibrosis, hepatic iron has also been linked to a lack of response to IFNα in chronic hepatitis C patients[44,45]. One mechanism by which this may occur was suggested by Di Bona and colleagues, who found that oxidative stress prevented IFNα induced phosphorylation of STAT-1 & 2 and subsequent upregulation of the antiviral proteins MxA and interferon regulatory factor 9, thereby impairing the antiviral action of IFNα[46].
The link between iron and fibrosis in chronic hepatitis C patients has, however, been challenged by Guyader and colleagues who looked at other confounding factors also known to influence both iron overload and fibrosis[47]. In their studies, liver iron was elevated in only 17% of patients, and correlated with age, male sex, and alcohol intake. They found no association between liver iron and fibrosis after adjusting for confounding variables, and suggested that iron should be considered more as a surrogate marker for disease severity rather than as a fibrogenic factor[47]. Iron is still further implicated in chronic hepatitis C progression by a number of groups investigating the effects of iron depletion on HCV-associated fibrosis. Kaito and colleagues demonstrated that phlebotomy treatment of HCV-infected patients alone was enough to reduce markers of liver damage (AST and ALT values), lipid peroxidation and oxidative stress[48]. In addition, patients treated by maintenance phlebotomy (keeping patients in a state of near iron deficiency with a serum ferritin level of 10 ng/mL) showed less inflammation and suppressed fibrosis progression[49]. Phlebotomy has also been used to increase the therapeutic effect of IFNα treatment of chronic hepatitis C by a number of groups[50,51]. How and why HCV should interfere with iron homeostasis is open to conjecture. However, there is some evidence to suggest that HCV infection may influence the expression of the iron homeostasis peptide, hepcidin. Nagashima and colleagues suggested that a failure in the regulation of serum prohepcidin levels (mediated by HCV) leads to elevated serum ferritin concentrations, and subsequently to progression of liver injury by iron overload in chronic hepatitis C patients[52]. Conversely, Aoki and colleagues found hepcidin mRNA expression in the liver did not correlate with AST, ALT levels, or viral load and no differences in hepcidin mRNA were found based on viral genotype or the presence of fibrosis[53].
MOLECULAR MECHANISMS OF IRON-INDUCED FIBROSIS
This section outlines the mechanisms by which iron is able to cause liver damage and the subsequent pathways activated that lead to hepatic fibrosis.
Role of iron in oxidative stress
In hepatocytes and KC, iron catalyses the production of hydroxyl radical (OH•) from reactive oxygen species (ROS), superoxide (O2•-) and hydrogen peroxide (H2O2) via a chemical reaction known as the Fenton reaction (Figure 1). ROS are the by-products of aerobic respiration reactions by cytochrome P450 (CYP) 2E1, and are also produced by the membrane-bound NADPH oxidase complex[54]. In addition, Fe catalyses the formation of NO2+ from peroxynitrite (ONOO-). ONOO- is formed when the cellular level of O2•- is elevated as this later reacts with nitric oxide (NO), produced by the constitutive and inducible nitric oxide synthase (NOS)[55,56] (Figure 1). Interestingly, nitric oxide has also a protective effect against oxidative stress, as it can inhibit lipid peroxidation and the generation of OH• by reacting with Fe3+[56,57]. NO2+ and OH• induce oxidative deterioration of biomolecules (lipid, protein and DNA), leading to tissue injury and cell death. In addition, iron can modulate gene expression in the cells leading to an alteration of cell function[58,59].
Figure 1.
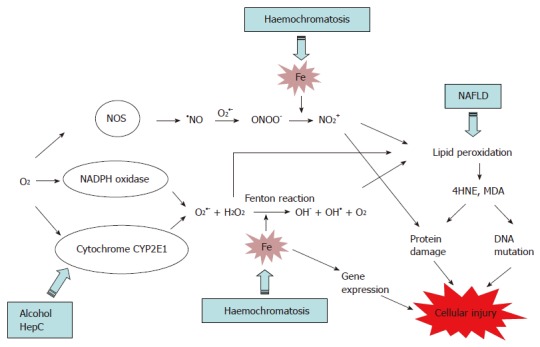
The involvement of iron in oxidative stress and its cytotoxic conse-quences. Iron catalyses the production of the reactive molecules OH• (via the Fenton reaction) and NO2+, which promote lipid peroxidation and protein damage leading to cellular injury. NOS, nitric oxide synthase; •NO, nitric oxide; ONOO-, peroxynitrite; O2•-, superoxide radical; OH•, hydroxyl radical; H2O2, hydrogen peroxide; NO2+, nitronium anion; Fe, iron; NAFLD, non-alcoholic fatty liver disease.
Alteration of essential biomolecules: NO2+ and OH• catalyse lipid peroxidation which is the process whereby electrons are transfered from the lipids in cell membranes to the free radical, resulting in cell damage. This process proceeds by a free radical chain reaction mechanism. It affects polyunsaturated fatty acids, as they contain multiple double bonds, between which lie methylene -CH2- groups that are especially susceptible to peroxidation. Lipid decomposition leads to the generation of thiobarbituric acid (TBA)-reactants and breakdown by-products 4-hydroxynonenal (4HNE) and malondialdehyde (MDA), which are used as marker of lipid peroxidation.
Lipid peroxidation affects the plasma membrane of the cell, but also increases the membrane fragility of a number of different cell organelles, such as lysosomes, which store excess iron, mitochondria and endoplasmic reticulum, leading to impaired cell funtion[2]. Lipid peroxidation of the mitochondrial membrane can lead to an increase in their permeability resulting in loss of the electrochemical gradient and release of the proapoptotic cytochrome C. This phenomenon is called mitochondria permeability transition. Damaged mitochondria generate yet more ROS that further enhance cell damage and activate proapoptotic signals[60].
Another consequence of lipid peroxidation is the damage of DNA and proteins, as lipid peroxidation products such as 4HNE and MDA can react with DNA bases[61] and the ε-NH2 group of lysine and histidine residues[62]. The presence of acetaldehyde, resulting from ethanol oxidation, increases the binding of MDA and its own binding to proteins in a synergistic manner, generating new hybrid adducts called MDA-acetaldehyde adducts[63]. These adducts may play a role in the development and progression of liver fibrosis as they have been shown to stimulate the secretion of several cytokines and chemokines by liver endothelial cells (TNFα, MCP-1, MIP-2, fibronectin) and HSC (MCP-1, MIP-2, uPA)[64-66].
Gene expression modulation: Iron or iron-induced oxidative stress have also been found to activate cell signalling cascades triggering apoptosis and necrosis pathway via NF-κB and AP-1 pathways respectively[60]. NF-κB promotes the synthesis and release of cytotoxic, proinflammatory and fibrogenic factors such as TNFα, IL-6 and MIP-1 that alter KC and hepatocyte function, and trigger HSC activation[58,59]. In HSC, AP-1 transcription factors are involved in the regulation of procollagen (I)[56]. In addition, AP-1 and NF-κB-dependent gene products modulate hepatocyte death induced by oxidative stress[67].
Regulation of intracellular levels of ROS: In the normal liver, hepatocytes are able to remove or neutralize oxidative molecules via enzymatic and non-enzymatic antioxidant processes, thereby maintaining a safe cellular level of ROS. For example gluthatione (GSH) is a tripeptide that neutralizes free radicals and ROS directly via a chemical reaction or via enzymatic reactions involving gluthatione-reductase or gluthatione-peroxidase[68]. Antioxidant agents such as vitamin A, C and E are also able to impair lipid peroxidation by breaking the chain reaction. Other antioxidants can act as inhibitors of CYP2E1, such as diallylsulfide[56]. However, when cellular defences are overwhelmed, there is an accumulation of ROS, which trigger cellular damage and apoptosis and, when this occurs in the liver, it can lead to fibrosis.
Iron driven oxidative stress promotes the development of hepatic fibrosis: Iron from the diet is absorbed into the enterocyte, and is either stored bound to ferritin or exported out into the plasma coupled to transferrin. In the liver, iron is taken up mainly by hepatocytes, and secondarily by KC. Hepatic iron uptake is mediated via HFE and the transferrin receptor, and iron is stored within the storage protein, ferritin[69]. Macrophages phagocytose senescent blood cells, and recycle the iron back to plasma. In iron-loading disorders, although hepatocytes remain the dominant site of iron deposition, KC can also store iron[70]. As mentioned earlier, the interplay between hepatocytes, KC and HSC and other extracellular proteins in the liver is important in the progression of hepatic fibrosis, and can be influenced by iron. Iron, as a key player in the oxidative reaction, may contribute directly and indirectly to HSC activation although the precise pathways remain unclear (Figure 2).
Figure 2.
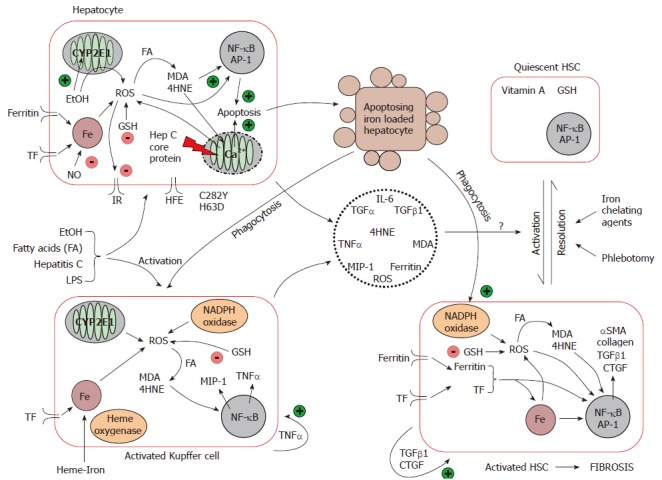
Mechanisms of iron-induced liver injury and fibrosis. Iron catalyses the formation of several reactive oxygen species in hepatocytes. Under normal circumstances, hepatocytes are able to effectively cope with oxidant stress. When the liver is subjected to a secondary insult that enhances hepatic oxidant stress, hepatic fibrosis begins to develop. Increased oxidative stress and other pathological modes of action of HCV, ethanol and steatosis, lead to mitochondrial dysfunction and hepatocyte apoptosis. Kupffer cell activation is achieved by phagocytosis of apoptosing hepatocytes in conjunction with the direct effects of iron, HCV infection, ethanol and steatosis on Kupffer cells. The concomitant hepatocyte damage/apoptosis and Kupffer cell activation is able to drive and maintain hepatic stellate cell activation leading to fibrosis and ultimately cirrhosis if left unchecked. Fe, iron; TF, transferrin; ROS, reactive oxygen species; FA, fatty acids; IR, insulin receptor; GSH, glutathione; TNFα, tumour necrosis factor α; NO, nitric oxide; IL-6, interleukin 6; TGFβ1, transforming growth factor β1; CTGF, connective tissue growth factor; αSMA, α smooth muscle actin; MDA, malondialdehyde; 4HNE, 4-hydroxynonenal; MIP-1, macrophage inflammatory protein-1; TGFα, transforming growth factor α; NF-κB, nuclear factor κB; AP-1, activating protein-1.
Indirect activation of HSC: Hepatocytes and KC are the main cells where iron–induced oxidative damage occurred as described earlier, i.e. lipid peroxidation and activation of proapoptotic/necrotic signalling. It is proposed that HSC activation occurs as a result of soluble factors (TGF-α/β1, TNFα, MIP-1, IL-6) produced by injured hepatocytes or KC following oxidative stress[71-73]. Indeed, the gene expression of two markers of HSC activation, collagen typeIand αSMA, is increased in studies where HSC are incubated with conditioned medium from iron loaded hepatocytes[72,74]. Collagen upregulation was also observed when HSC were co-cultured with hepatocytes expressing CYP2E1, which is the principal cytochrome involved in the oxidation of ethanol when the concentration of ethanol is elevated[74]. This effect was even more pronounced in the presence of iron, and was prevented by several antioxidants suggesting a oxidation mediated upregulation of these profibrogenic genes[74].
KC have been shown to play a role in HSC activation. When KC and HSC are co-cultured, HSC proliferation is increased along with HSC expression of αSMA and collagen typeI, when compared to the culture of HSC alone[73]. Friedman and Arthur demonstrated that KC conditioned medium activated HSC and stimulated HSC proliferation[75]. After exposure to several stimuli such as LPS, ethanol, fatty acid, HCV infection or the phagocytosis of injured hepatocytes, KC become activated and produced ROS via PKC dependant activation of NADPH oxidase and via the CYP1E2[55,59,76,77]. Deugnier and colleagues observed KC containing phagocytosed hepatocellular debris in hemochromatosis liver tissue[78]. Tsukamoto and colleagues demonstrated that haeme-derived iron primed hepatic macrophages for NF-κB activation, and enhanced the expression of the pro-inflammatory genes TNFα and MIP-1[79]. TNFα is known to play a key role in promoting KC activation[80], and also prevents the HSC from undergoing apoptosis[81] thereby promoting fibrosis.
In addition to the production of profibrogenic and proinflammatory cytokines that activate HSC, KC release iron-loaded tissue ferritin that may interact with the ferritin receptor on activated HSC[82]. Furthermore, hepatocyte and KC-generated ROS can be released from the cell, and enhance the perpetuation of HSC activation[83]. In contrast, the role of the toxic by-products of lipid peroxidation such as MDA and 4HNE has been studied by Olynyk and others who showed that these compounds did not directly activate HSC[84,85].
Direct activation of HSC: Activated HSC express a specific receptor for H-ferritin[86] which appears to regulate the expression of αSMA[82]. Ruddell and colleagues have preliminary evidence which suggest that ferritin upregulates genes involved in HSC activation via a PKCζ dependent pathway[87]. In addition, activated HSC are also known to express a transferrin receptor which can enhance the expression of αSMA and collagen type I within these cells[88]. The role of free iron in the induction of HSC activation has been studied by Gardi and colleagues who demonstrated that iron can stimulate type I collagen gene expression but this was not mediated by lipid peroxidation[89]. This is supported by other studies which rule out the involvement of intracellular lipid peroxidation in the activation of HSC[84,85,90].
However, in other studies, intracellular oxidative stress has been shown to induce collagen expression in HSC, and that HSC lipid peroxidation can be triggered by hepatocytes[91,92]. A recent study by Zhan and colleagues supports the role of lipid peroxidation in HSC in the upregulation of procollagen expression[93]. They showed that HSC are able to phagocytose apoptotic bodies (i.e. injured hepatocytes), which triggers the activation of the stellate cells NADPH oxidase and its production of superoxide within and outside the cell. They also showed that the upregulation of procollagen I following the phagocytosis of apoptotic bodies was NADPH oxidase-dependent.
In conclusion, iron may have a direct effect on the activation of HSC or via the modulation of intracellular oxidative stress. However, this may be a minor role in the activation of the HSC when you consider that hepatocytes are the main cells that take up iron, and that quiescent HSC do not express the transferrin or ferritin receptors[86,88]. The inherent ability of iron to catalyse the production ROS in hepatocytes and KC appears likely the main mechanism by which it is able to influence hepatic disease progression. Iron, in conjunction with other factors, may overwhelm the liver in terms of its innate ability to cope with oxidative stress thereby causing disease.
Iron and other oxidation-related molecules
As described in Figure 1, iron is a key player in oxidative reactions that produced toxic ROS. However, the cellular level of other molecules, such as H2O2 and unsaturated lipids, also influence the degree of oxidation that occurs in the cells and thereby the degree of fibrosis. Indeed, the role of iron along with alcohol, steatosis and HCV in exacerbating liver injury has been the subject of fairly intensive study. The effects of feeding rats a diet supplemented with iron and alcohol was found to enhance hepatic localisation of MDA and 4HNE as well as increase serum levels of ALT and AST to two-fold that of rats fed ethanol alone[94]. Both liver tissues and HSC isolated from rats fed ethanol and iron were also found to express elevated levels of procollagenα1 (I) and TGFβ1 mRNA compared to those fed ethanol alone[94]. Kato and colleagues also suggested TGFα derived from alcohol exposed hepatocytes may also contribute to hepatic fibrosis in ALD[95]. Ethanol is known to upregulate the expression of CYP2E1[74] which would exacerbate the potential oxidative stress exerted by iron in hepatocytes due to the catalysis of more ROS. In the same manner, accumulation of fatty acid within the cells will increase oxidative damage due to the enhanced peroxidation of lipid.
HCV alone has been shown to increase ROS generation and also lipid peroxidation in hepatocytes[96]. It does this via the core protein, which is able to alter mitochondrial Ca2+ uptake, and also it induces endoplasmic reticulum (ER) stress and enhances ER to mitochondria Ca2+ transfer[97]. Iron overload is also able to induce mitochondrial dysfunction (as visualised by ultrastructural alterations), and in mice over expressing the HCV core polyprotein, iron increases the risk of those mice going on to develop HCC[98].
CONCLUSION
Iron plays an integral part in the progression of hepatic fibrosis, and it does this via its ability to catalyse the formation of highly reactive and damaging ROS. The damage done by ROS includes lipid peroxidation, protein and DNA modification leading ultimately to apoptosis and necrosis. However, the literature also indicates that co-factors such as steatosis, ethanol and HCV infection contribute to iron-induced hepatic injury. Antioxidant therapies have been tested in clinical trials with limited success with regards to the prevention and reversal of liver fibrosis. Iron depletion therapies have also been used with varying degrees of success to treat all four diseases of the liver outlined in this review. Continued investigation into the molecular mechanisms of iron toxicity, and how it leads to hepatic fibrosis may give a better understanding of the role of iron as a cofactor in the progression of liver disease leading ultimately to novel anti-fibrotic therapies.
Footnotes
Supported by NHMRC Program Grant 339400
Co-first-authors: Richard G Ruddell
S- Editor Liu Y L- Editor Negro F E- Editor Ma WH
References
- 1.Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol. 2005;202:199–211. doi: 10.1016/j.taap.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 2.Ramm GA, Ruddell RG. Hepatotoxicity of iron overload: mechanisms of iron-induced hepatic fibrogenesis. Semin Liver Dis. 2005;25:433–449. doi: 10.1055/s-2005-923315. [DOI] [PubMed] [Google Scholar]
- 3.Bertrand-Philippe M, Ruddell RG, Arthur MJ, Thomas J, Mungalsingh N, Mann DA. Regulation of tissue inhibitor of metalloproteinase 1 gene transcription by RUNX1 and RUNX2. J Biol Chem. 2004;279:24530–24539. doi: 10.1074/jbc.M311804200. [DOI] [PubMed] [Google Scholar]
- 4.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neubauer K, Saile B, Ramadori G. Liver fibrosis and altered matrix synthesis. Can J Gastroenterol. 2001;15:187–193. doi: 10.1155/2001/870205. [DOI] [PubMed] [Google Scholar]
- 6.Sato M, Suzuki S, Senoo H. Hepatic stellate cells: unique characteristics in cell biology and phenotype. Cell Struct Funct. 2003;28:105–112. doi: 10.1247/csf.28.105. [DOI] [PubMed] [Google Scholar]
- 7.Ruddell RG, Oakley F, Hussain Z, Yeung I, Bryan-Lluka LJ, Ramm GA, Mann DA. A role for serotonin (5-HT) in hepatic stellate cell function and liver fibrosis. Am J Pathol. 2006;169:861–876. doi: 10.2353/ajpath.2006.050767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alcolado R, Arthur MJ, Iredale JP. Pathogenesis of liver fibrosis. Clin Sci (Lond) 1997;92:103–112. doi: 10.1042/cs0920103. [DOI] [PubMed] [Google Scholar]
- 9.Marra F, Choudhury GG, Pinzani M, Abboud HE. Regulation of platelet-derived growth factor secretion and gene expression in human liver fat-storing cells. Gastroenterology. 1994;107:1110–1117. doi: 10.1016/0016-5085(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 10.Iredale JP. Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis. 2001;21:427–436. doi: 10.1055/s-2001-17557. [DOI] [PubMed] [Google Scholar]
- 11.Niederau C, Fischer R, Sonnenberg A, Stremmel W, Trampisch HJ, Strohmeyer G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med. 1985;313:1256–1262. doi: 10.1056/NEJM198511143132004. [DOI] [PubMed] [Google Scholar]
- 12.Ramm GA, Crawford DH, Powell LW, Walker NI, Fletcher LM, Halliday JW. Hepatic stellate cell activation in genetic haemochromatosis. Lobular distribution, effect of increasing hepatic iron and response to phlebotomy. J Hepatol. 1997;26:584–592. doi: 10.1016/s0168-8278(97)80424-2. [DOI] [PubMed] [Google Scholar]
- 13.Adams PC. Is there a threshold of hepatic iron concentration that leads to cirrhosis in C282Y hemochromatosis? Am J Gastroenterol. 2001;96:567–569. doi: 10.1111/j.1572-0241.2001.03472.x. [DOI] [PubMed] [Google Scholar]
- 14.Powell LW, Dixon JL, Ramm GA, Purdie DM, Lincoln DJ, Anderson GJ, Subramaniam VN, Hewett DG, Searle JW, Fletcher LM, et al. Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch Intern Med. 2006;166:294–301. doi: 10.1001/archinte.166.3.294. [DOI] [PubMed] [Google Scholar]
- 15.Bassett ML, Halliday JW, Powell LW. Value of hepatic iron measurements in early hemochromatosis and determination of the critical iron level associated with fibrosis. Hepatology. 1986;6:24–29. doi: 10.1002/hep.1840060106. [DOI] [PubMed] [Google Scholar]
- 16.Falize L, Guillygomarc'h A, Perrin M, Lainé F, Guyader D, Brissot P, Turlin B, Deugnier Y. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases. Hepatology. 2006;44:472–477. doi: 10.1002/hep.21260. [DOI] [PubMed] [Google Scholar]
- 17.Fletcher LM, Powell LW. Hemochromatosis and alcoholic liver disease. Alcohol. 2003;30:131–136. doi: 10.1016/s0741-8329(03)00128-9. [DOI] [PubMed] [Google Scholar]
- 18.Piperno A, Fargion S, D'Alba R, Roffi L, Fracanzani AL, Vecchi L, Failla M, Fiorelli G. Liver damage in Italian patients with hereditary hemochromatosis is highly influenced by hepatitis B and C virus infection. J Hepatol. 1992;16:364–368. doi: 10.1016/s0168-8278(05)80671-3. [DOI] [PubMed] [Google Scholar]
- 19.Powell EE, Ali A, Clouston AD, Dixon JL, Lincoln DJ, Purdie DM, Fletcher LM, Powell LW, Jonsson JR. Steatosis is a cofactor in liver injury in hemochromatosis. Gastroenterology. 2005;129:1937–1943. doi: 10.1053/j.gastro.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 20.Booth ML, Chey T, Wake M, Norton K, Hesketh K, Dollman J, Robertson I. Change in the prevalence of overweight and obesity among young Australians, 1969-1997. Am J Clin Nutr. 2003;77:29–36. doi: 10.1093/ajcn/77.1.29. [DOI] [PubMed] [Google Scholar]
- 21.Alla V, Bonkovsky HL. Iron in nonhemochromatotic liver disorders. Semin Liver Dis. 2005;25:461–472. doi: 10.1055/s-2005-923317. [DOI] [PubMed] [Google Scholar]
- 22.George DK, Goldwurm S, MacDonald GA, Cowley LL, Walker NI, Ward PJ, Jazwinska EC, Powell LW. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology. 1998;114:311–318. doi: 10.1016/s0016-5085(98)70482-2. [DOI] [PubMed] [Google Scholar]
- 23.Bonkovsky HL, Jawaid Q, Tortorelli K, LeClair P, Cobb J, Lambrecht RW, Banner BF. Non-alcoholic steatohepatitis and iron: increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J Hepatol. 1999;31:421–429. doi: 10.1016/s0168-8278(99)80032-4. [DOI] [PubMed] [Google Scholar]
- 24.Mendler MH, Turlin B, Moirand R, Jouanolle AM, Sapey T, Guyader D, Le Gall JY, Brissot P, David V, Deugnier Y. Insulin resistance-associated hepatic iron overload. Gastroenterology. 1999;117:1155–1163. doi: 10.1016/s0016-5085(99)70401-4. [DOI] [PubMed] [Google Scholar]
- 25.Festi D, Colecchia A, Sacco T, Bondi M, Roda E, Marchesini G. Hepatic steatosis in obese patients: clinical aspects and prognostic significance. Obes Rev. 2004;5:27–42. doi: 10.1111/j.1467-789x.2004.00126.x. [DOI] [PubMed] [Google Scholar]
- 26.Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology. 1999;30:1356–1362. doi: 10.1002/hep.510300604. [DOI] [PubMed] [Google Scholar]
- 27.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 28.Younossi ZM, Gramlich T, Bacon BR, Matteoni CA, Boparai N, O'Neill R, McCullough AJ. Hepatic iron and nonalcoholic fatty liver disease. Hepatology. 1999;30:847–850. doi: 10.1002/hep.510300407. [DOI] [PubMed] [Google Scholar]
- 29.Facchini FS, Hua NW, Stoohs RA. Effect of iron depletion in carbohydrate-intolerant patients with clinical evidence of nonalcoholic fatty liver disease. Gastroenterology. 2002;122:931–939. doi: 10.1053/gast.2002.32403. [DOI] [PubMed] [Google Scholar]
- 30.Chapman RW, Morgan MY, Laulicht M, Hoffbrand AV, Sherlock S. Hepatic iron stores and markers of iron overload in alcoholics and patients with idiopathic hemochromatosis. Dig Dis Sci. 1982;27:909–916. doi: 10.1007/BF01316575. [DOI] [PubMed] [Google Scholar]
- 31.Pascoe A, Kerlin P, Steadman C, Clouston A, Jones D, Powell L, Jazwinska E, Lynch S, Strong R. Spur cell anaemia and hepatic iron stores in patients with alcoholic liver disease undergoing orthotopic liver transplantation. Gut. 1999;45:301–305. doi: 10.1136/gut.45.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raynard B, Balian A, Fallik D, Capron F, Bedossa P, Chaput JC, Naveau S. Risk factors of fibrosis in alcohol-induced liver disease. Hepatology. 2002;35:635–638. doi: 10.1053/jhep.2002.31782. [DOI] [PubMed] [Google Scholar]
- 33.Ganne-Carrié N, Christidis C, Chastang C, Ziol M, Chapel F, Imbert-Bismut F, Trinchet JC, Guettier C, Beaugrand M. Liver iron is predictive of death in alcoholic cirrhosis: a multivariate study of 229 consecutive patients with alcoholic and/or hepatitis C virus cirrhosis: a prospective follow up study. Gut. 2000;46:277–282. doi: 10.1136/gut.46.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brittenham GM. Iron chelators and iron toxicity. Alcohol. 2003;30:151–158. doi: 10.1016/s0741-8329(03)00101-0. [DOI] [PubMed] [Google Scholar]
- 35.Bridle K, Cheung TK, Murphy T, Walters M, Anderson G, Crawford DG, Fletcher LM. Hepcidin is down-regulated in alcoholic liver injury: implications for the pathogenesis of alcoholic liver disease. Alcohol Clin Exp Res. 2006;30:106–112. doi: 10.1111/j.1530-0277.2006.00002.x. [DOI] [PubMed] [Google Scholar]
- 36.Lim CY, Kowdley KV. Optimal duration of therapy in HBV-related cirrhosis. J Antimicrob Chemother. 2007;60:2–6. doi: 10.1093/jac/dkm102. [DOI] [PubMed] [Google Scholar]
- 37.Seeff LB. The natural history of chronic hepatitis C virus infection. Clin Liver Dis. 1997;1:587–602. doi: 10.1016/s1089-3261(05)70323-8. [DOI] [PubMed] [Google Scholar]
- 38.Blumberg BS, Lustbader ED, Whitford PL. Changes in serum iron levels due to infection with hepatitis B virus. Proc Natl Acad Sci USA. 1981;78:3222–3224. doi: 10.1073/pnas.78.5.3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Bisceglie AM, Axiotis CA, Hoofnagle JH, Bacon BR. Measurements of iron status in patients with chronic hepatitis. Gastroenterology. 1992;102:2108–2113. doi: 10.1016/0016-5085(92)90339-z. [DOI] [PubMed] [Google Scholar]
- 40.Piperno A, D'Alba R, Roffi L, Pozzi M, Farina A, Vecchi L, Fiorelli G. Hepatitis C virus infection in patients with idiopathic hemochromatosis (IH) and porphyria cutanea tarda (PCT) Arch Virol Suppl. 1992;4:215–216. doi: 10.1007/978-3-7091-5633-9_46. [DOI] [PubMed] [Google Scholar]
- 41.Ikura Y, Morimoto H, Johmura H, Fukui M, Sakurai M. Relationship between hepatic iron deposits and response to interferon in chronic hepatitis C. Am J Gastroenterol. 1996;91:1367–1373. [PubMed] [Google Scholar]
- 42.Martinelli AL, Ramalho LN, Zucoloto S. Hepatic stellate cells in hepatitis C patients: relationship with liver iron deposits and severity of liver disease. J Gastroenterol Hepatol. 2004;19:91–98. doi: 10.1111/j.1440-1746.2004.03255.x. [DOI] [PubMed] [Google Scholar]
- 43.Rigamonti C, Andorno S, Maduli E, Morelli S, Pittau S, Nicosia G, Boldorini R, Sartori M. Iron, hepatic stellate cells and fibrosis in chronic hepatitis C. Eur J Clin Invest. 2002;32 Suppl 1:28–35. doi: 10.1046/j.1365-2362.2002.0320s1028.x. [DOI] [PubMed] [Google Scholar]
- 44.Olynyk JK, Reddy KR, Di Bisceglie AM, Jeffers LJ, Parker TI, Radick JL, Schiff ER, Bacon BR. Hepatic iron concentration as a predictor of response to interferon alfa therapy in chronic hepatitis C. Gastroenterology. 1995;108:1104–1109. doi: 10.1016/0016-5085(95)90209-0. [DOI] [PubMed] [Google Scholar]
- 45.Piperno A, Sampietro M, D'Alba R, Roffi L, Fargion S, Parma S, Nicoli C, Corbetta N, Pozzi M, Arosio V, et al. Iron stores, response to alpha-interferon therapy, and effects of iron depletion in chronic hepatitis C. Liver. 1996;16:248–254. doi: 10.1111/j.1600-0676.1996.tb00737.x. [DOI] [PubMed] [Google Scholar]
- 46.Di Bona D, Cippitelli M, Fionda C, Cammà C, Licata A, Santoni A, Craxì A. Oxidative stress inhibits IFN-alpha-induced antiviral gene expression by blocking the JAK-STAT pathway. J Hepatol. 2006;45:271–279. doi: 10.1016/j.jhep.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 47.Guyader D, Thirouard AS, Erdtmann L, Rakba N, Jacquelinet S, Danielou H, Perrin M, Jouanolle AM, Brissot P, Deugnier Y. Liver iron is a surrogate marker of severe fibrosis in chronic hepatitis C. J Hepatol. 2007;46:587–595. doi: 10.1016/j.jhep.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 48.Kaito M, Iwasa M, Kobayashi Y, Fujita N, Tanaka H, Gabazza EC, Adachi Y, Kojima Y, Nakagawa N, Watanabe S. Iron reduction therapy by phlebotomy reduces lipid peroxidation and oxidative stress in patients with chronic hepatitis C. J Gastroenterol. 2006;41:921–922. doi: 10.1007/s00535-006-1871-5. [DOI] [PubMed] [Google Scholar]
- 49.Yano M, Hayashi H, Wakusawa S, Sanae F, Takikawa T, Shiono Y, Arao M, Ukai K, Ito H, Watanabe K, et al. Long term effects of phlebotomy on biochemical and histological parameters of chronic hepatitis C. Am J Gastroenterol. 2002;97:133–137. doi: 10.1111/j.1572-0241.2002.05436.x. [DOI] [PubMed] [Google Scholar]
- 50.Fontana RJ, Israel J, LeClair P, Banner BF, Tortorelli K, Grace N, Levine RA, Fiarman G, Thiim M, Tavill AS, et al. Iron reduction before and during interferon therapy of chronic hepatitis C: results of a multicenter, randomized, controlled trial. Hepatology. 2000;31:730–736. doi: 10.1002/hep.510310325. [DOI] [PubMed] [Google Scholar]
- 51.Tsai NC, Zuckerman E, Han SH, Goad K, Redeker AG, Fong TL. Effect of iron depletion on long-term response to interferon-alpha in patients with chronic hepatitis C who previously did not respond to interferon therapy. Am J Gastroenterol. 1997;92:1831–1834. [PubMed] [Google Scholar]
- 52.Nagashima M, Kudo M, Chung H, Ishikawa E, Hagiwara S, Nakatani T, Dote K. Regulatory failure of serum prohepcidin levels in patients with hepatitis C. Hepatol Res. 2006;36:288–293. doi: 10.1016/j.hepres.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 53.Aoki CA, Rossaro L, Ramsamooj R, Brandhagen D, Burritt MF, Bowlus CL. Liver hepcidin mRNA correlates with iron stores, but not inflammation, in patients with chronic hepatitis C. J Clin Gastroenterol. 2005;39:71–74. [PubMed] [Google Scholar]
- 54.Hampton MB, Fadeel B, Orrenius S. Redox regulation of the caspases during apoptosis. Ann N Y Acad Sci. 1998;854:328–335. doi: 10.1111/j.1749-6632.1998.tb09913.x. [DOI] [PubMed] [Google Scholar]
- 55.Videla LA, Fernández V, Tapia G, Varela P. Oxidative stress-mediated hepatotoxicity of iron and copper: role of Kupffer cells. Biometals. 2003;16:103–111. doi: 10.1023/a:1020707811707. [DOI] [PubMed] [Google Scholar]
- 56.Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35:297–306. doi: 10.1016/s0168-8278(01)00142-8. [DOI] [PubMed] [Google Scholar]
- 57.Sergent O, Griffon B, Morel I, Chevanne M, Dubos MP, Cillard P, Cillard J. Effect of nitric oxide on iron-mediated oxidative stress in primary rat hepatocyte culture. Hepatology. 1997;25:122–127. doi: 10.1002/hep.510250123. [DOI] [PubMed] [Google Scholar]
- 58.Lin M, Rippe RA, Niemelä O, Brittenham G, Tsukamoto H. Role of iron in NF-kappa B activation and cytokine gene expression by rat hepatic macrophages. Am J Physiol. 1997;272:G1355–G1364. doi: 10.1152/ajpgi.1997.272.6.G1355. [DOI] [PubMed] [Google Scholar]
- 59.Cao Q, Mak KM, Lieber CS. Cytochrome P4502E1 primes macrophages to increase TNF-alpha production in response to lipopolysaccharide. Am J Physiol Gastrointest Liver Physiol. 2005;289:G95–107. doi: 10.1152/ajpgi.00383.2004. [DOI] [PubMed] [Google Scholar]
- 60.Czaja MJ. Induction and regulation of hepatocyte apoptosis by oxidative stress. Antioxid Redox Signal. 2002;4:759–767. doi: 10.1089/152308602760598909. [DOI] [PubMed] [Google Scholar]
- 61.Bartsch H, Nair J. Oxidative stress and lipid peroxidation-derived DNA-lesions in inflammation driven carcinogenesis. Cancer Detect Prev. 2004;28:385–391. doi: 10.1016/j.cdp.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 62.Uchida K, Szweda LI, Chae HZ, Stadtman ER. Immunochemical detection of 4-hydroxynonenal protein adducts in oxidized hepatocytes. Proc Natl Acad Sci USA. 1993;90:8742–8746. doi: 10.1073/pnas.90.18.8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tuma DJ. Role of malondialdehyde-acetaldehyde adducts in liver injury. Free Radic Biol Med. 2002;32:303–308. doi: 10.1016/s0891-5849(01)00742-0. [DOI] [PubMed] [Google Scholar]
- 64.Thiele GM, Worrall S, Tuma DJ, Klassen LW, Wyatt TA, Nagata N. The chemistry and biological effects of malondialdehyde-acetaldehyde adducts. Alcohol Clin Exp Res. 2001;25:218S–224S. doi: 10.1097/00000374-200105051-00035. [DOI] [PubMed] [Google Scholar]
- 65.Kharbanda KK, Todero SL, Shubert KA, Sorrell MF, Tuma DJ. Malondialdehyde-acetaldehyde-protein adducts increase secretion of chemokines by rat hepatic stellate cells. Alcohol. 2001;25:123–128. doi: 10.1016/s0741-8329(01)00174-4. [DOI] [PubMed] [Google Scholar]
- 66.Kharbanda KK, Shubert KA, Wyatt TA, Sorrell MF, Tuma DJ. Effect of malondialdehyde-acetaldehyde-protein adducts on the protein kinase C-dependent secretion of urokinase-type plasminogen activator in hepatic stellate cells. Biochem Pharmacol. 2002;63:553–562. doi: 10.1016/s0006-2952(01)00883-8. [DOI] [PubMed] [Google Scholar]
- 67.Luedde T, Trautwein C. Intracellular survival pathways in the liver. Liver Int. 2006;26:1163–1174. doi: 10.1111/j.1478-3231.2006.01366.x. [DOI] [PubMed] [Google Scholar]
- 68.Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND. Glutathione Metabolism and Its Implications for Health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 69.Sharma N, Butterworth J, Cooper BT, Tselepis C, Iqbal TH. The emerging role of the liver in iron metabolism. Am J Gastroenterol. 2005;100:201–206. doi: 10.1111/j.1572-0241.2005.40152.x. [DOI] [PubMed] [Google Scholar]
- 70.Halliday JW, Searle J. Hepatic iron deposition in human disease and animal models. Biometals. 1996;9:205–209. doi: 10.1007/BF00144626. [DOI] [PubMed] [Google Scholar]
- 71.Britton RS, Ramm GA, Olynyk J, Singh R, O'Neill R, Bacon BR. Pathophysiology of iron toxicity. Adv Exp Med Biol. 1994;356:239–253. doi: 10.1007/978-1-4615-2554-7_26. [DOI] [PubMed] [Google Scholar]
- 72.Parkes JG, Templeton DM. Modulation of stellate cell proliferation and gene expression by rat hepatocytes: effect of toxic iron overload. Toxicol Lett. 2003;144:225–233. doi: 10.1016/s0378-4274(03)00222-4. [DOI] [PubMed] [Google Scholar]
- 73.Nieto N. Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] Kupffer cells on stellate cells. Hepatology. 2006;44:1487–1501. doi: 10.1002/hep.21427. [DOI] [PubMed] [Google Scholar]
- 74.Wu D, Cederbaum AI. Oxidative stress mediated toxicity exerted by ethanol-inducible CYP2E1. Toxicol Appl Pharmacol. 2005;207:70–76. doi: 10.1016/j.taap.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 75.Friedman SL, Arthur MJ. Activation of cultured rat hepatic lipocytes by Kupffer cell conditioned medium. Direct enhancement of matrix synthesis and stimulation of cell proliferation via induction of platelet-derived growth factor receptors. J Clin Invest. 1989;84:1780–1785. doi: 10.1172/JCI114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Falcón V, Acosta-Rivero N, Shibayama M, Chinea G, Gavilondo JV, de la Rosa MC, Menéndez I, Gra B, Dueñas-Carrera S, Viña A, et al. HCV core protein localizes in the nuclei of nonparenchymal liver cells from chronically HCV-infected patients. Biochem Biophys Res Commun. 2005;329:1320–1328. doi: 10.1016/j.bbrc.2005.02.107. [DOI] [PubMed] [Google Scholar]
- 77.Rusyn I, Bradham CA, Cohn L, Schoonhoven R, Swenberg JA, Brenner DA, Thurman RG. Corn oil rapidly activates nuclear factor-kappaB in hepatic Kupffer cells by oxidant-dependent mechanisms. Carcinogenesis. 1999;20:2095–2100. doi: 10.1093/carcin/20.11.2095. [DOI] [PubMed] [Google Scholar]
- 78.Deugnier YM, Loréal O, Turlin B, Guyader D, Jouanolle H, Moirand R, Jacquelinet C, Brissot P. Liver pathology in genetic hemochromatosis: a review of 135 homozygous cases and their bioclinical correlations. Gastroenterology. 1992;102:2050–2059. doi: 10.1016/0016-5085(92)90331-r. [DOI] [PubMed] [Google Scholar]
- 79.Tsukamoto H, Lin M, Ohata M, Giulivi C, French SW, Brittenham G. Iron primes hepatic macrophages for NF-kappaB activation in alcoholic liver injury. Am J Physiol. 1999;277:G1240–G1250. doi: 10.1152/ajpgi.1999.277.6.G1240. [DOI] [PubMed] [Google Scholar]
- 80.Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, Kitamura N, Toda K, Kaneko T, Horie Y, et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut. 2006;55:415–424. doi: 10.1136/gut.2005.071118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saile B, Matthes N, Knittel T, Ramadori G. Transforming growth factor beta and tumor necrosis factor alpha inhibit both apoptosis and proliferation of activated rat hepatic stellate cells. Hepatology. 1999;30:196–202. doi: 10.1002/hep.510300144. [DOI] [PubMed] [Google Scholar]
- 82.Ramm GA, Britton RS, O'Neill R, Kohn HD, Bacon BR. Rat liver ferritin selectively inhibits expression of alpha-smooth muscle actin in cultured rat lipocytes. Am J Physiol. 1996;270:G370–G375. doi: 10.1152/ajpgi.1996.270.2.G370. [DOI] [PubMed] [Google Scholar]
- 83.Nieto N, Friedman SL, Cederbaum AI. Cytochrome P450 2E1-derived reactive oxygen species mediate paracrine stimulation of collagen I protein synthesis by hepatic stellate cells. J Biol Chem. 2002;277:9853–9864. doi: 10.1074/jbc.M110506200. [DOI] [PubMed] [Google Scholar]
- 84.Montosi G, Garuti C, Martinelli S, Pietrangelo A. Hepatic stellate cells are not subjected to oxidant stress during iron-induced fibrogenesis in rodents. Hepatology. 1998;27:1611–1622. doi: 10.1002/hep.510270622. [DOI] [PubMed] [Google Scholar]
- 85.Olynyk JK, Khan NA, Ramm GA, Brown KE, O'Neill R, Britton RS, Bacon BR. Aldehydic products of lipid peroxidation do not directly activate rat hepatic stellate cells. J Gastroenterol Hepatol. 2002;17:785–790. doi: 10.1046/j.1440-1746.2002.02798.x. [DOI] [PubMed] [Google Scholar]
- 86.Ramm GA, Britton RS, O'Neill R, Bacon BR. Identification and characterization of a receptor for tissue ferritin on activated rat lipocytes. J Clin Invest. 1994;94:9–15. doi: 10.1172/JCI117353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ruddell RG, Rutherford PS, Barwood JM, Hoang-Le D, Santambrogio P, Arosio P, Ramm GA. Ferritin upregulates the expression of NF-κB-dependant genes associated with hepatic stellate cell activation via PI3-kinase, PKCζ and MAPK. Hepatology. 2004;40:A269. [Google Scholar]
- 88.Bridle KR, Crawford DH, Ramm GA. Identification and characterization of the hepatic stellate cell transferrin receptor. Am J Pathol. 2003;162:1661–1667. doi: 10.1016/S0002-9440(10)64300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gardi C, Arezzini B, Fortino V, Comporti M. Effect of free iron on collagen synthesis, cell proliferation and MMP-2 expression in rat hepatic stellate cells. Biochem Pharmacol. 2002;64:1139–1145. doi: 10.1016/s0006-2952(02)01257-1. [DOI] [PubMed] [Google Scholar]
- 90.Maher JJ, Neuschwander-Tetri BA. Manipulation of glutathione stores in rat hepatic stellate cells does not alter collagen synthesis. Hepatology. 1997;26:618–623. doi: 10.1002/hep.510260313. [DOI] [PubMed] [Google Scholar]
- 91.Bedossa P, Houglum K, Trautwein C, Holstege A, Chojkier M. Stimulation of collagen alpha 1(I) gene expression is associated with lipid peroxidation in hepatocellular injury: a link to tissue fibrosis? Hepatology. 1994;19:1262–1271. [PubMed] [Google Scholar]
- 92.Nieto N, Friedman SL, Greenwel P, Cederbaum AI. CYP2E1-mediated oxidative stress induces collagen type I expression in rat hepatic stellate cells. Hepatology. 1999;30:987–996. doi: 10.1002/hep.510300433. [DOI] [PubMed] [Google Scholar]
- 93.Zhan SS, Jiang JX, Wu J, Halsted C, Friedman SL, Zern MA, Torok NJ. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43:435–443. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 94.Tsukamoto H, Horne W, Kamimura S, Niemelä O, Parkkila S, Ylä-Herttuala S, Brittenham GM. Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest. 1995;96:620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kato J, Sato Y, Inui N, Nakano Y, Takimoto R, Takada K, Kobune M, Kuroiwa G, Miyake S, Kohgo Y, et al. Ethanol induces transforming growth factor-alpha expression in hepatocytes, leading to stimulation of collagen synthesis by hepatic stellate cells. Alcohol Clin Exp Res. 2003;27:58S–63S. doi: 10.1097/01.ALC.0000078614.44983.97. [DOI] [PubMed] [Google Scholar]
- 96.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, Weinman SA. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 97.Wang T, Weinman SA. Causes and consequences of mitochondrial reactive oxygen species generation in hepatitis C. J Gastroenterol Hepatol. 2006;21 Suppl 3:S34–S37. doi: 10.1111/j.1440-1746.2006.04591.x. [DOI] [PubMed] [Google Scholar]
- 98.Furutani T, Hino K, Okuda M, Gondo T, Nishina S, Kitase A, Korenaga M, Xiao SY, Weinman SA, Lemon SM, et al. Hepatic iron overload induces hepatocellular carcinoma in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology. 2006;130:2087–2098. doi: 10.1053/j.gastro.2006.02.060. [DOI] [PubMed] [Google Scholar]