

Abstract
The features of JAK-STAT signaling in liver cells are discussed in the current review. The role of this signaling cascade in carcinogenesis is accentuated. The possible involvement of this pathway and alteration of its elements are compared for normal cholangiocytes, cholangiocarcinoma predisposition and development. Prolactin and interleukin-6 are described in detail as the best studied examples. In addition, the non-classical nuclear translocation of cytokine receptors is discussed in terms of its possible implication to cholangiocarcinoma development.
Keywords: Cholangiocarcinoma, Janus tyrosine Kinases, Signal Transducers and Activators of Transcription, Prolactin, Interleukin-6, Cytokine receptors, Receptor tyrosine kinases
INTRODUCTION
The signaling pathway of Janus tyrosine Kinases-Signal Transducers and Activators of Transcription (JAK-STAT) is activated by a variety of hormones (prolactin, growth hormone, leptin, erythropoietin), as well as cytokines, and growth factors via their receptors. JAK-STAT signaling participates in the regulation of cell proliferation, differentiation, survival, motility, and apoptosis in different organs including liver[1]. During the last decades, the data on cytokine/growth factor receptors expression and functions in different liver cell types (hepatocytes, cholangiocytes, Kupffer and stellate cells) have rapidly grown. It highlights the importance of JAK-STAT signaling in normal liver physiology and pathology. Moreover, deregulation of this pathway is closely associated with tumorigenesis.
Cholangiocarcinoma is a malignancy of the biliary epithelium with the worst prognosis among other liver tumors. It is relatively rare, but it frequently progressively increases. Molecular cholangiocarcinoma markers are mainly nonspecific and require further investigation. The main risk factors for cholangiocarcinoma development are hyperplasia of bile duct epithelia induced by bile duct obstruction, hepatolithiasis, inflammation, fibrosis, and some other factors[2-4]. Though the role of the JAK-STAT cascade is poorly deciphered both in normal cholangiocytes and in cholangiocarcinoma cells, a number of papers have been published recently where the role of prolactin (Prl), growth hormone (GH) and interleukin-6 (IL-6) and expression of corresponding receptors in cholangiocytes has been shown[5-7]. Expression of these receptors undergoes essential changes under hyperplasia of bile duct epithelia induced by bile duct obstruction, thus the role of the JAK-STAT pathway should attract further investigations in this area.
PRINCIPLES OF JAK-STAT SIGNALING
The main site for JAK-STAT signaling initiation is the cellular membrane; however, internalization pathways have been recently shown to be also important for the generation of comprehensive biological effect and sustaining signal transduction. Among other intracellular targets of receptor-JAK-STAT the cascade nucleus plays a key role[8-10]. Mitochondrial targeting of some cytokine receptors (GH receptor) has also been described[11].
The initial steps of signaling cascade activation by hormone (cytokine) are still intensively studied, but for many of them an interaction between signaling molecules and preformed receptor dimers leads to rotation of intercellular parts of receptor and subsequent activation of receptor-associated JAKs[12] followed by STAT docking on receptor and its phosphorylation, dimerization, and nuclear translocation (Figure 1).
Figure 1.
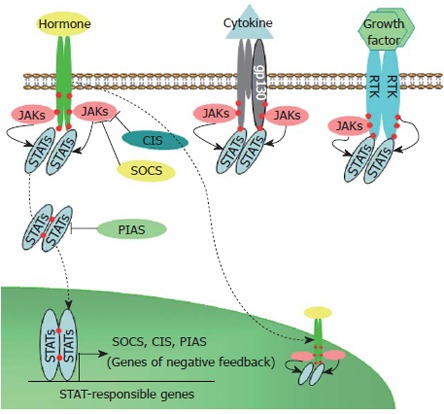
Schematic representation of JAK-STAT signaling induced by hormones, cytokines, and growth factors. RTK: Receptors tyrosine kinases; JAK: Jannus kinase; STA: Signal transducer and activator of transcription; SOCS: Suppressor of cytokine signaling; CIS: Cytokine induced suppressor; PIAS: Protein inhibitors of activated STATs. Solid arrows - phosphorylation; Dashed arrows - nuclear translocation; Blunt arrows - inhibition; Red circles - phosphorylated tyrosines.
Different JAKs (JAK1, JAK2, JAK3, and Tyk2) and STATs (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6) participate in JAK-STAT signal transduction. STAT3 and STAT5 are expressed in many cell types, activated by various hormones, cytokines and growth factors and play a role in different biological responses, whereas other STAT proteins (STAT1, 2, 4 and 6) are revealed mainly in specific cell types and participate predominantly in host defense mechanisms[13,14]. Phosphorylated STAT dimers can bind to STAT-responsive elements in the promoters of various genes, resulting in modulation of their transcription. STATs often act on gene transcription in cooperation or competition with NF-κB, AP-1, and a glucocorticoid receptor due to close co-localization of correspondent binding sites[15-17] accomplishing a crosstalk with other signaling cascades.
Suppressors of cytokine signaling (SOCS) proteins are able to inhibit JAK-STAT signaling. SOCS1 and SOCS3 are the most potent and broadly distributed suppressors with similar inhibition effects on gp130 related, Prl, and GH signaling. Promoter regions of SOCS1 and SOCS3 genes possess functional STAT binding elements and activation of JAK-STAT signaling induces rapid up-regulation of SOCS proteins by STAT-dependent pathways. In accordance with negative feedback mechanism JAK-STAT signaling is inhibited by SOCS proteins association with catalytic domains of JAKs as well as by SOCS binding to phosphorylated tyrosine residues of cytokine, GH, Prl, leptin, and erythropoietin (Epo) receptors which act as docking sites for downstream signaling[18-20]. STAT activation can also be inhibited by direct interaction with protein inhibitors of activated STAT proteins (PIAS)[21].
In some cases activation of STATs occurs indepen-dently of JAKs and is involved in receptor tyrosine kinase signaling. This activation is induced by direct interaction with receptor tyrosine kinases or by subsequent downstream signaling with implication of other kinases such as Src. For example, STAT3 is activated in response to stimulation by EGF, HGF, CSF-1, PDGF and other growth factors; and their corresponding receptor tyrosine kinases (ErbB1, ErbB-2, c-met, CSF-1, PDGF receptors, and others) possess a common STAT3 docking motif in their cytoplasmic domains. STAT5a phosphorylation is induced by EGF[13,22,23].
JAK-STAT SIGNALING AND CARCINOGENESIS
Altered activation of JAK-STAT signaling has been shown in multiple solid tumors and leukemia[24-26]. In different experimental models tumorigenesis is associated with increased expression and/or activity of JAK1, 2[27,28] or STAT3, 5[29]. A constitutively active mutant of STAT3 has been shown to transform rat fibroblasts[30], and the increased expression of wild type STAT5 in the lymphoid lineage induced T cell leukemia in mice[31]. A constitutively active mutant of JAK2 has been identified in patients with polycythaemia vera[32]. Moreover, increased levels of correspondent ligands can be associated with JAK-STAT induced carcinogenesis: in animal models of prostate and mammary gland hyperplasia with local overexpression of Prl[33,34]; in humans hyperprolactinemia is considered as a risk factor for breast and, probably, prostate cancer[35,36]; overexpression of GH in transgenic mice leads to increased frequency of mammary adenocarcinoma[37]. A particular and essential role for neoplasia progression of autocrine production of Prl has been revealed[34,38]. Only autocrine, but not exocrine GH, is suggested to promote neoplasia[39-41].
The role of the JAK-STAT cascade is not limited to induction of carcinogenesis: for example, the predominant role of TYK2 and STAT1 in many types of cancer is antitumorigenic via regulation of apoptosis[42]. Moreover, in some models JAK1 and 2 also play an antitumorigenic role[43]. Prl induced activation of JAK2 prevents breast cancer cells mesenchymal transition and acts as an invasion suppressor[44,45]. Reduced phosphorylation of STAT5 in breast cancer highlights patients with a worse prognosis of disease development[46].
CHOLANGIOCARCINOMA DEVELOPMENT AND JAK-STAT SIGNALING
A number of signaling molecules acting via the JAK-STAT pathway participate in regulation of bile duct cell functions. Among them, Prl and IL-6 (activation of complete JAK-STAT cascade), and EGF, HGF (activation of STAT via receptor tyrosine kinases) have their receptors in human and animal cholangiocarcinoma tissues and cell lines. Other JAK-STAT signaling molecules involved in the development of other cancer types have not been yet investigated.
Since the development of cholangiocarcinoma is closely related with obstructive, fibrotic, cirrhotic, and inflammation changes of biliary epithelium leading to its hyperplasia or dysplasia, it is reasonable to compare the peculiarity of JAK-STAT signaling induced by different ligands in normal cholangiocytes, bile duct cells proliferating under conditions of different hepatic pathologies considered as a predisposition to cholangiocarcinoma development, and in cholangiocarcinoma cells.
HORMONAL INDUCTION OF JAK-STAT SIGNALING AND CHOLANGIOCARCINOMA DEVELOPMENT
Prolactin signaling
Prl is a multifunctional pituitary hormone that also works as a locally produced cytokine. Prl regulates differentiation, proliferation, water-salt balance in different cell types including many ductal structures (such as ductal systems of mammary, lacrimal, submaxillary, prostate glands, pancreas, and kidney) and possesses high immunomodulatory activity[47,48]. Prl participates in the stimulation of rat bile duct cell proliferation and cholangiocyte regulation of bile water salt balance[49,50].
Prl receptor (PrlR) exists in several isoforms with a tissue specific ratio of expression. Long and short rat PrlR isoforms are the result of alternative splicing of cytoplasmic domain exons of a single gene. Several intermediate PrlR isoforms with varying cytoplasmic domains have been found in human normal and tumor tissues. The short PrlR isoform has been suggested to negatively regulate activity of the long isoform by heterodimerization. JAK-STAT pathway with predominant usage of JAK2 and STAT5 proteins is the main signaling pathway activated by Prl. STAT1 and STAT3 proteins may also be involved in Prl signaling with nuclear translocation of their homo- and heterodimers. SOCS1, 2, and 3 attenuate Prl signaling[47,48,51-53].
Prolactin JAK-STAT signaling and their alterations under conditions of cholangiocarcinoma predisposition and development
Systemic Prl levels are elevated under different liver pathologies associated with increased cholangiocarcinoma incidents[54-56], while local Prl production by the cholangiocarcinoma cell line (RS-1) is observed only in a few samples (Ostroukhova & Smirnova, unpublished observations).
Normal animal and human bile duct cells express PrlR messenger RNA and protein at a relatively low level. Contrary to hepatocytes where short PrlR isoform predominates, normal rat bile duct cells express a predominantly long receptor isoform that is similar for such Prl target tissues, such as the uterus, pituitary, and mammary gland[7,57].
Hyperplasia of bile duct epithelia in rats induced by bile duct obstruction or normally occurring in immature animals as a risk factor of cholangiocarcinoma development is characterized by sharp elevation of cholangiocyte PrlR expression in both males and females. Similarly, obstructive human liver diseases with jaundice development and enrichment of bile ducts are accompanied by a pronounced increase in cholangiocyte PrlR expression as compared with mild cholestasis without jaundice with limited affect on bile duct cell proliferating activity[57]. Obstructive conditions induce both dramatic elevation of the long receptor isoform and an increase in short isoform expression in bile duct cells. The level and isoform ratio of cholangiocyte PrlR in normal and obstructive conditions are only slightly dependent on gonadal hormones and independent of Prl[7,49,57].
Human cholangiocarcinoma samples are characterized by high PrlR expression significantly exceeding PrlR expression in cholangiocytes and hepatocytes under conditions of non-tumor liver diseases (Ostroukhova & Smirnova, unpublished observations). Intrahepatic transplantation of RS1 cholangiocarcinoma cells induces significant elevation of tumor cell PrlR expression (Figure 2) as compared both with subcutaneous tumor transplants and cholangiocytes of normal animals[58].
Figure 2.
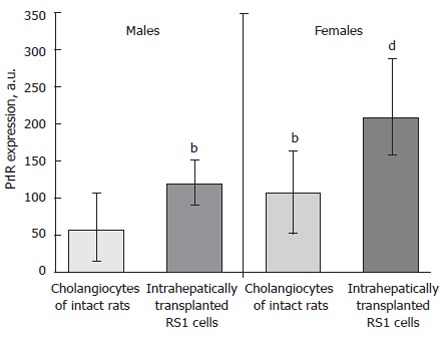
Increasing of prolactin receptor expression in rat liver transplanted RS-1 cholangiocarcinoma cell line. a.u, arbitrary units of intensity of PrlR expression. Medians, upper and lower quartiles. bP < 0.001, vs intact males, dP < 0.001, vs intact females.
In cholangiocarcinoma cells the isoforms ratio is similar to the ratio observed in cholangiocytes proliferating under obstructive conditions. Surprisingly, relatively high levels of PrlR in transplantable cholangiocarcinoma have been revealed not only in female recipients, but also in male counterparts under conditions of relatively low levels of both Prl and estrogens[58]. Thus, it seems possible that the main regulators of PrlR expression in cholangiocarcinoma differ from sex hormones and may be connected with tumor derived autocrine/paracrine factors. Such unusual regulation may be an early event of cholangiocyte dysplasia since sharp elevation of PrlR expression in both males and females primarily occurs in bile duct cells proliferating in response to bile duct obstruction[7,49].
Prolactin - estrogen cross-talk and cholangiocarcinoma development
Prl as a female sex hormone acts in close connection with estrogens. In addition to systemic positive regulation of Prl production by estrogens and vise versa, a cross-talk between these hormones has been shown at the level of target tissues.
Prl stimulates both alpha and beta mRNA estrogen receptor expression in some tissues and putative STAT5α and STAT5β response elements have been identified in their promoters[59].
Estradiol via estrogen receptor (ER) alpha activates transcriptional expression of human PrlR through promoter III that functions in different tissues including liver. Estrogens increase and tamoxifen reduces expression of Prl receptor mRNA in breast tumors and some other tissues[60-62]. Estrogens participate in regulation of downstream Prl signaling using different mechanisms. Estrogens can attenuate JAK-STAT signaling by Prl. Estrogens via ER α up-regulate SOCS2 expression thus diminishing JAK-STAT signaling. ER alpha activation could diminish PrlR signaling via direct interaction with STAT5α which leads to reduced STAT5α nuclear translocation. On the contrary, nongenomic action of estrogens that requires a ligand-binding domain of the membrane estrogen receptor alpha leads to rapid induction of STAT3 and STAT5 phosphorylation, their nuclear translocation, and transcriptional activity[61,63-66].
All these facts are very important taking into account that both PrlR and ERs are sharply increased in human cholangiocarcinoma samples and cell lines. While normal cholangiocytes express extremely low ERs, cholangiocarcinoma cells are characterized by intensive cytoplasmic and nuclear expression of ERα and ERβ as well as increased ERα/ERβ ratio that is typical for other cancer types. Besides, these estrogens potentiate and tamoxifen attenuates proliferation of human cholangiocarcinoma cell lines in vitro[4,6]. As in the case of PrlR, high level of ER expression in cholangiocarcinoma have been revealed not only in females, but also in males[67]. Based on both cytoplasmic and nuclear ER localization in cholangiocarcinoma cells it seems possible that estrogen regulation of Prl induced tumor JAK-STAT signaling is both nuclear and nongenomic and its bottom-line effects should be investigated carefully. It is interesting that in spite of high sensitivity of cholangiocarcinoma cells to both types of female sex hormones cholangiocarcinoma development is more frequent in men (1.5-1) than in women[68]. It may be due to attenuation of Prl tumor signaling by estrogens in the case of relatively high levels of both hormones in women. In any case combined therapy with tamoxifen and bromocriptine may be perspective clinical strategy for treatment of cholangiocarcinoma.
Cross-talk of Prl with EGF signaling leading to ErbB1 and ErbB2 phosphorylation has also been found, e.g. in breast cancer cells[69]. Since both Prl receptor and ERbB1 and ErbB2 are present in cholangiocarcinoma cells it may be relevant also to cholangiocarcinoma Prl signaling.
Growth hormone (GH) JAK-STAT signaling
As in the case of Prl, JAK-STAT signaling activated by GH mainly includes JAK2 activation and STAT5 phosphorylation, though STAT1 and 3 activation are sometimes also implicated. GH induces the expression of several negative regulators of GH signaling: SOCS1, 2, and 3, CIS[70,71].
GH receptor is present in normal rat cholangiocytes and isolated cholangiocytes respond to GH stimulation by IGF-1 secretion that in turn promotes cholangiocyt proliferation. GH receptor expression is increased in rat cholangiocytes after common bile duct ligation[72]. Contrary, in total liver of mice under the same conditions and humans with obstructive cholestasis and cirrhosis, changes of GH receptor expression and IGF-1 production are opposite[73]. GH signaling in cholangiocarcinoma cells has not been yet investigated.
Leptin JAK-STAT signaling
Leptin receptor is homologous to gp130 and can activate JAK2, mainly STAT3, but also STAT5, STAT6, STAT1 proteins[74-77]. SOCS3 is a negative regulator of leptin JAK-STAT signaling[18,78,79].
Autocrine/paracrine leptin production and leptin mRNA have been shown in different types of cancer cell lines in vitro and in some tumors in vivo. Serum leptin level also increases in patients with some cancer types[76,80]. Leptin is considered an agent responsible for overcoming the antiproliferative effect of antiestrogen therapy in different cancer types. Leptin receptors have been found in different cancer types and cell lines and leptin signaling is associated with suppression of tumor cell apoptosis[13,78].
Leptin expression has been detected in liver stellate cells and its local production is increased under obstructive cholestasis conditions. Leptin acts as a direct hepatic stellate cell survival agent[81-83]. Serum leptin is also elevated after common bile duct ligation and CCl4 induced liver fibrosis that are both risk factors for cholangiocarcinoma development[84]. The leptin receptor has been found in bile duct cells of one type of mammalian species[85]. The role of leptin induction of JAK-STAT signaling needs to be investigated in cholangiocarcinoma development, and known STAT3 activation during cholangiocarcinoma progression (see below) may be partly due to leptin signaling.
Erythropoietin (Epo) JAK-STAT signaling
Epo is produced mainly by the fetal liver and the adult kidney. Epo interaction with Epo receptor activates JAK2 and STAT5[86,87]. Due to its erythropoietic functions, Epo has been widely used for prevention and treatment of cancer-associated anemia. However, Epo proangiogenic activity can compromise its beneficial effects in cancer patients.
Expression of Epo and Epo receptors has been found in a number of nonhematopoietic tissues as well as in different types of primary tumors and tumor cell lines suggesting the generation of autocrine-paracrine growth stimulatory Epo-Epo receptor loop in cancer cells. Experimental blockade of Epo signaling results in inhibition of different tumor growth and angiogenesis with concomitant elevation of apoptotic death of tumor and vascular endothelial cells[87-90].
Epo and/or Epo receptor expression have been found in liver stromal cells and some primary hepatic tumors, and tumor cell lines as well as in Kupffer cells during regeneration[87,89-93]. Still, we have not found any data on Epo expression and signaling in human cholangiocarcinoma and its animal models. Based on the frequency of Epo and Epo receptor expression in different types of tumor cells, a key role of the fetal liver in Epo production, a role of hepatic progenitor cells, enriched in fetal liver, in liver cancer development as well as possibility of bone marrow derived liver stem cells to differentiate into biliary cells[90,94-97] it is easy to assume that the same situation occurs in cholangiocarcinoma development. Several facts support this assumption. In an earlier study the elevation of liver Epo production after bile duct ligation has been shown[98]. Data on the possibility of tissue specific estrogen modulation of local Epo expression together with elevation of estrogen receptor expression in human cholangiocarcinoma are also of great interest in this connection[6,86,99,100].
CYTOKINE INDUCTION OF JAK-STAT SIGNALING AND CHOLANGIOCARCINOMA DEVELOPMENT
Interleukine-6 (IL-6) induced JAK-STAT signaling
IL-6 and other cytokines of this family have their own receptors, but need a common receptor subunit gp130 for signal transduction. Binding of IL-6 to its receptor leads to dimerization of gp130, resulting in activation of gp130-associated JAK kinases (JAK1, JAK2, and TYK2) and subsequent activation of STAT3 proteins forming homodimers or rarely heterodimers with STAT1. There is reciprocal negative regulation of JAK-STAT3 and MAPK signaling by IL-6[15,101,102]. SOCS3 activated by STAT3 suppresses IL-6 signaling[15,18-20].
Alterations of local and systemic IL-6 level under conditions of cholangiocarcinoma predisposition and development
Autocrine/paracrine production of IL-6 by normal liver bile duct cells as well as by cultured human intrahepatic biliary epithelial cells is very low. Bile duct obstruction triggers pronounced elevation of IL-6 mRNA and protein production by bile duct cells, which can be mediated by inflammatory conditions[103-105]. A pronounced autocrine/paracrine secretion of IL-6 by different human intrahepatic cholangiocarcinoma cell lines has been observed in vitro[106,107]. Obstructive changes as well as cholangiocarcinoma development are also associated with elevation of systemic plasma IL-6 level[108].
Alterations of IL-6 signaling under conditions of cholangiocarcinoma predisposition and development
In vitro normal bile duct cells express both IL-6 receptor and gp130 messenger RNA and protein, but their expression level is further increased in cholangiocarcinoma cells. Appearance of autocrine IL-6 production and high level of its receptors lead to autocrine/paracrine loop of IL-6 signaling during cholangiocarcinoma progression[5,109].
IL-6 target genes in cholangiocarcinoma progression
IL-6 enhances cell survival and proliferation of human malignant cholangiocytes by up-regulation of myeloid cell leukemia-1 (mcl-1) mRNA and protein expression which is a key member of the antiapoptotic Bcl-2 protein family[107,110]. Disruption of IL-6 signaling at different levels diminishes Mcl-1 promoter activity, its mRNA and protein level in human cholangiocarcinoma cell lines.
Sustained IL-6/JAK-STAT signaling and enhanced Mcl-1 expressions in cholangiocarcinoma are associated with at least partial epigenetic SOCS3 silencing. In human cholangiocarcinoma tissues and cell lines unusual inverse correlation exists between phosphorylated STAT-3 and SOCS3 protein expression. SOCS3 negative cholangiocarcinoma (as well as cholangiocarcinoma lines) samples are characterized by extensive methylation of SOCS3 promoter as compared with nontumor tissue. Treatment with demethylating agent restores IL-6 induction of SOCS3 leading to termination of phosphorylated STAT3 response, reduction of cholangiocarcinoma cells level of Mcl-1, and sensitization of cholangiocarcinoma cells to Tumor necrosis factor Related Apoptosis Inducing Ligand (TRAIL)-mediated apoptosis[111,112].
GROWTH FACTOR INDUCTION OF STAT SIGNALING AND CHOLANGIOCARCINOMA DEVELOPMENT
HGF induced STAT signaling
HGF is a growth factor acting via receptor tyrosine kinase c-Met and inducing matrix dissociation, motility of epithelial cells, and enhancing invasiveness of tumor cells. In the 5′-flanking region of human HGF gene cis-acting STAT element has been found. Interruption of STAT3 signaling by dominant-negative STAT3 reduces HGF promoter activity[113,114].
Interaction of HGF with its receptor c-Met leads to receptor dimerization and phosphorylation of intracellular receptor domain which forms the docking sites for interaction with multiple downstream signaling molecules including STAT3. STAT3 activation has been shown to be necessary for full biological responses of HGF and HGF induced proliferation[115-117]. In hepatocytes and some other cell types HGF activates the STAT3 signaling pathway, moreover, not only STAT3 but also STAT1α/STAT1β and STAT5 interact with c-Met[118,119]. STAT3 phosphorylation induced by HGF/c-Met signaling is limited by consequent positive HGF action on SOCS3 expression in some cell types. SOCS3 has been observed to act as a negative regulator of HGF-induced cell migration[120-122].
Alterations of local and systemic HGF level
In situ hybridization studies have demonstrated that there is a close paracrine ligand-receptor relationship between HGF expressed in mesenchymal cells and c-Met expressed in the adjacent epithelial cells. HGF production is stimulated by various cytokines including IL-6-type cytokines. Both HGF and c-met mRNAs and protein products are overexpressed in cholestatic animal liver and human hepatolithiasis specimens[123-125]. Serum HGF level is elevated in cholangiocarcinoma patients[126]. Cholangiocarcinoma cell lines do not secrete HGF, but their co-cultivation with neutrophils induces a high level of HGF secretion by these mesenchymal cells and may enhance invasiveness of tumor cells[127].
Alterations of HGF signaling
c-Met is expressed in cultured mouse biliary epithelial cells and liver bile ducts[123,128]. c-Met activation and elevation of c-met mRNA and protein expression have been shown in mouse chemically induced cholangiocarcinoma, different cholangiocarcinoma cell lines, and human cholangiocarcinoma patients. c-Met overexpression is more prominent in induced cholangiocarcinoma in rats and human cholangiocarcinoma cell lines as compared to normal and hyperplastic intrahepatic biliary epithelium[3,5,126,129-131]. The presence of HGF-c-Met-STAT3 positive autocrine/paracrine loop in cholangiocarcinoma may confer increased survival, growth, and invasiveness during cholangiocarcinoma progression. The malignancy of cholangiocarcinoma cells containing activated STAT3 may be at least partly due to stimulation of HGF expression in adjacent mesenchymal cells and c-met overexpression in tumor cells.
ERbB2 induced JAK-STAT and STAT signaling
ErbB2 also known as HER-2 or HER-2/neu is a member of ErbB receptor tyrosine kinase family acting mainly as the most common partner for heteromerization for other members of this receptor family (ErbB1, ErbB3 and ERbB2) among which EGF is the main ligand for ErbB1 (EGFR). ErbB receptors may be constitutively associated with JAK kinases. Direct association between ErbB2 and other ErbB receptors with STAT3 has also been found. Src kinases are the other molecules for ErbB receptor mediated STAT signaling and potential upstream regulators of JAK kinases. Ligand induced receptor dimerization leads to recruitment of Src kinase that induces JAK and STAT3 phosphorylation, dimerization and nuclear translocation[132,133]. Neuregulin induced heterodimers of ErbB2 and ErbB4 may activate STAT5[23]. Expression of SOCS1, 3, 4, 5 is elevated by EGF treatment[134-136]. Tumor cell types that over express ErbB2 preferentially activate prosurvival STAT signaling. In different tumors an autocrine loop between cytokines and ErbB receptors induces constitutive STAT3 activation[133,137].
In human non-neoplastic hepatic tissue ErbB2 mRNA and protein products have been found only in large mature bile ducts or have not been revealed at all[138-140]. Human gallstone disease and common bile duct ligation in rats are accompanied by appearance or elevation of ErbB2 expression in cholangiocytes as compared with normal liver but its expression was lower than in cases of cholangiocarcinoma[138,141,142]. ErbB2 overexpression has been revealed in different human biliary tract cancer cell lines, xenobiotic induced rodent cholangiocarcinoma, as well as in human cholangiocarcinoma tissue samples. Overexpression of both ErbB1 (EGFR) and ErbB2 may serve as a base for enhancement of EGF signaling in cholangiocarcinoma. Altered ERbB2 expression occurs early in cholangiocarcinogenesis and may play an important role in its progression[129,130,139,143,144].
PERSPECTIVE THERAPEUTIC ADDRESSING OF THE JAK-STAT PATHWAY
As illustrated above, signaling of most hormones, cytokines and growth factors involved in cholangiocyte regulation seems to be enhanced under the progression of cholangiocarcinoma. It is associated, not only with increment of their systemic levels, but also (probably mainly) due to their local hepatic secretion and increment of the expression of their receptors. Taking together these observations we can suggest that local manipulation of JAK-STAT activation can be a very perspective therapeutic approach for managing of cholangiocarcinoma at different stages of its progression. Recent advances in development of hormonal and cytokine antagonists (recombinant proteins for Prl and GH and peptide for IL-6[145-147]) give us an efficient tool for this treatment. Since synthetic specific inhibitors of JAK and other tyrosine kinases are intensively developed, we could suggest their efficient application for managing of cholangiocarinoma in addition to antagonists.
For the moment, the most specific and efficient way to inhibit the components of the JAK-STAT pathway is local expression (tissue-specific) of dominant negative analogues of JAKs and STATs. Evidently, their application is now limited to laboratory investigations and needs further development of genetic therapy. Nevertheless, it is a potent tool for precise pre-clinical investigations of JAK-STAT inhibition in cholangiocarcinoma. Thus, in the next chapter we focus on a detailed description of the main players of the JAK-STAT pathway in cholangiocarcinoma.
JAK-STAT SIGNALING UNDER CONDITIONS OF CHOLANGIOCARCINOMA PREDISPOSITION AND DEVELOPMENT
Presented data indicate that a number of receptors associated with JAK-STAT signaling are expressed in normal cholangiocytes, their expression increases under conditions of cholangiocarcinoma predisposition and is additionally elevated in cholangiocarcinoma cells (Table 1). Preferably used components of JAK-STAT signaling for such receptors are shown in Table 2. Thus, STAT5 and STAT3 proteins are the main downstream components for hormones, growth factors, and cytokines using JAK-STAT signaling in normal, proliferating, and malignant cholangiocytes and both STAT3 and STAT5 are implicated in promotion of cell survival in a variety of other normal and cancer tissues. SOCS3 is the primary negative regulator of cholangiocyte JAK-STAT signaling (Table 2). Among all these components, only STAT3 and SOCS3 have been investigated in cholangiocarcinoma cells.
Table 1.
Cholangiocyte receptors associated with the JAK-STAT signaling pathways: alterations of expression during cholangiocarcinoma predisposition and development
| Receptor type | Norma |
Direction of alteration |
References | |
| Cholangiocarcinoma predisposition | Cholangiocarcinoma development | |||
| Prolactin receptor | Low | Up | Up Up | [7,58,148] |
| IL-6 receptor/gp130 | Present | Up | Up Up | [2,5] |
| ErbB-2 | Low/Absent | Up | Up Up | [2,138-140] |
| c-Met | Present | Up | Up Up | [2,5] |
| Growth hormone receptor | Moderate | Up | ? | [72] |
| Leptin receptor | Present | ? | ? | [85] |
Table 2.
Known components of JAK-STAT signaling for receptors expressed in cholangiocytes
| Receptor type | JAK | STATs | Negative regulators of JAK-STAT signaling | References |
| Prolactin receptor | JAK2 | STAT5 > STAT3 > STAT1 | SOCS3, SOCS1, SOCS2, CIS | [47,48,51-53] |
| Growth hormone receptor | JAK2 | STAT5 > STAT3 > STAT1 | SOCS3, SOCS1, SOCS2, CIS | [70,71] |
| Leptin receptor | JAK2 | STAT3 > STAT5, STAT6, STAT1 | SOCS3 | [74-79] |
| IL-6 receptor/gp130 | JAK1, 2, Tyk2 | STAT3 > STAT1 | SOCS3 | [15,18-20,101,102,110-112] |
| ErbB-2 | No, JAK3, TYK2 | STAT3 > STAT5, STAT6 | SOCS3, SOCS1, SOCS4, SOCS5 | [23,132-136,149,150] |
| c-Met | No | STAT3 > STAT5, STAT1 | SOCS3 | [115-122] |
STAT3
STAT3 plays an important role in transduction of survival signals downstream of both JAKs and receptor tyrosine kinases. Aberrant and frequently constitutive activation of STAT3 has been described in a variety of human cancers. Activation of STAT3 in tumors is associated also with tumor escape from immune attack[151,152].
In normal liver phosphorylated STAT3 localizes in epithelial cells lining large bile ducts and peribiliary glands[104]. STAT3 was constitutively phosphorylated in tested cholangiocarcinoma cell lines but not in nonmalignant cholangiocytes. Mcl-1 promotor has STAT3 binding site. STAT3 site-directed mutagenesis decreases Mcl-1 promotor activity[111].
SOCS3
STATs activation is negatively regulated by SOCS proteins. SOCSs are generally considered as a kind of tumor suppressor gene. In some hepatocarcinoma cell lines aberrant methylation in STAT-binding sites of the SOCS3 gene has been associated with loss of SOCS3 function and enhancement of tumor cell growth and migration[153]. Epigenetic inactivation of tumor suppressor genes by hypermethylation is relevant to cholangiocarcinoma development[154]. Known sustained STAT3 activation during cholangiocarcinoma progression may be partly due to SOCS3 epigenetic silencing due to hypermethylation of its promoter in human cholangiocarcinoma tissues and cell lines. Inverse correlation has been observed between phosphorylated STAT3 and SOCS3 protein expression in cholangiocarcinoma specimens. In cholangiocarcinoma samples failing to express SOCS3, extensive methylation of SOCS3 promoter has been shown in comparison with paired nontumor tissue. Methylation of SOCS3 promoter is also identified in two cholangiocarcinoma cell lines. Treatment with demethylating agent leads to restoration of SOCS3 expression, subsequent termination of STAT activation, and reduction of cellular levels of Mcl-1[112].
Cholangiocarcinoma markers and STAT-responsible genes
Based on the data on molecular alterations in human cholangiocarcinoma reviewed by Sirica, 2005, we have tried to analyze whether cholangiocarcinoma marker genes have putative STAT response elements in their promoters. As shown in Table 3 a number of genes involved in cholangiocarcinoma growth signaling, cell cycle regulation, and apoptosis inhibition possess candidate STAT3-, STAT5-, and STAT-1-binding sites in their promoters. Prl and IL-6 induced STAT5 and STAT3 activation is essential for up-regulation of their transcription activity in different tumor types and leads mainly to tumor progression[2,16,86,107,110-112,155,156].
Table 3.
Cholangiocarcinoma marker genes with promoter putative STAT responsible elements
| Cholangiocarcinoma altered genes1 | Direction of alteration1 | Putative STAT responsible elements | Regulation by STAT | References |
| HGF | Up | STAT3 | Up | [113,114] |
| VEGF | Up | STAT3 | Up | [157] |
| COX-2 | Up | STAT3, STAT5 | Up | [158] |
| Cyclin D1 | Up | STAT5 | Up | [16,155] |
| p53 | Down and up | STAT1 | Up | [159] |
| p21waf1/cip1 | Down | STAT1, STAT3, STAT5 | Up | [155,160,161] |
| p27kip1 | Down | STAT3 | Up | [162] |
| Bcl-2 | Up | STAT3 | Up | [16] |
| Bcl-Xl | Up | STAT3, STAT5 | Up | [155,163] |
| Mcl-1 | Up | STAT3 | Up | [111,112] |
| MUC1 | Up | STAT3, STAT1 | Up | [164] |
| MUC4 | Up | STAT | Up | [165] |
Altered genes and direction of changes of their expression are adopted from[2].
We can assume that cholangiocarcinoma markers (presented in Table 3) including growth factors, members of pro-inflammatory signaling pathway, CDK inhibitors, cyclins, members of the antiapoptotic Bcl-2 protein family including Mcl-1, Bcl-2, Bcl-XL, may serve as final potential molecular targets of JAK-STAT cholangiocarcinoma signaling induced by Prl, IL-6, HGF, EGF and some other molecules acting via JAK-STAT signaling. These promoters STAT-binding sites may be involved in overexpression of these markers in cholangiocarcinoma cells.
It is important to note that down-regulation of CDK inhibitors (p21waf1/cip1, p27kip1) is a characteristic feature of cholangiocarcinoma development while activated STATs up-regulate them in some other tissues (Table 3), thus the role of JAK-STAT signaling cannot be restricted to progression cholangiocarcinoma.
PRIMARY NUCLEAR JAK-STAT SIGNALING AND CHOLANGIOCARCINOMA DEVELOPMENT
Internalization pathways have also been demonstrated for signaling molecules associated with the JAK-STAT pathway. Nuclear accumulation of such molecules, ligand dependent nuclear translocation of corresponding receptors, nuclear localization of JAKs, and primary nuclear activation of STAT proteins have been revealed in different cell types and experimental systems. While membrane JAK-STAT signaling is the main signaling pathway in normal conditions, the importance of the primary nuclear pathway of JAK-STAT signaling rises under conditions of cell proliferation including preneoplastic and neoplastic proliferation. Appearance of components of JAK-STAT signaling in the nucleus or the enrichment of their nuclear pool seems to be associated with their participation in the regulation of proliferative processes and may be related to cholangiocarcinoma progression.
Ligands (such as Prl, GH, leptin, IL-1, IL-5, IFN-γ, TGF-α) and receptors acting via the JAK-STAT pathway or associated with STAT signaling by other mechanisms have been found in the nuclear compartment of different cell types mainly under conditions of high proliferative status and cancer development[8,10,166-169].
Nuclear translocation of Prl bound to Prl receptors has been revealed after Prl stimulation of Nb2 Prl-dependent lymphoma cells and the human breast cancer cell line T47D. Another mechanism suggests that Prl enters the nucleus in complex with cyclophilin B. In the nucleus this complex interacts with STAT5 and potentiates its activity facilitating STAT5 DNA binding. Prl/cyclophilin B complex directly interacts with the STAT5 N-terminus and appears to induce dissociation of PIAS3 from its complex with STAT5[170,171].
Both GH and GH receptor are subject to rapid nuclear translocation. Rat growth hormone binding protein (GHBP) that is a product of alternative splicing of GH receptor gene with an intact extracellular domain and without transmembrane and intracellular domains replaced by short carboxyterminal sequences, is also localized in cell nuclei in in vitro and in vivo experiments upon GH stimulation. Nuclear localized GHBP acts as a potent enhancer of STAT5 mediated transcription presumably by binding to PIAS proteins and thereby enhances the action of both GH and other members of the cytokine receptor superfamily[9,172].
Receptor tyrosine kinases associated with the JAK-STAT pathway or STAT signaling have also been found in the nuclear compartment. Nuclear EGF receptor (ErbB1) has been detected in highly proliferative tissues, such as regenerating liver, specimens of primary tumor, and cancer cell lines. Expression of nuclear ErbB1 correlates positively with increased level of cyclin D1 and negatively with overall survival in breast cancer patients. Nuclear ErbB1 co-localizes and interacts with importins α1β1, carriers that are critical for macromolecule nuclear import[8,168,169,173-176]. ErbB1 might act as a transcription factor to activate genes required for high proliferating activity. After EGF stimulation and ErbB1 nuclear translocation EGF-ErbB1 complex associates with cyclin D1 promoter element designated AT-rich sequence (RTRS)[177].
The ability of nucleus translocation is also inherent for other receptors of the ErbB family (ErbB2, ErbB3, and ErbB4). Rat p185neu, ErbB2, and ErbB3 receptors exist in the nucleus as full length molecules, while ErbB4 may undergo γ-secretase-mediated cleavage and with nuclear translocation of C-terminal 80-kDa fragment that represents a soluble cytoplasmic domain with an active tyrosine kinase region[10,178]. Nuclear ErbB2 associates with a specific nucleotide sequence (named HER-2-associated sequence - HAS) of the COX-2 gene promoter and is able to activate its transcription[168,173,175-178].
The hypothesis of a primary nuclear JAK-STAT signaling pathway is supported by data on constitutive expression of JAK1 and JAK2 in the cell nucleus. JAK1 and JAK2 molecules have been shown to contain nuclear localization sequences and both kinases have been revealed in nuclei of rat hepatocytes, pancreatic islet cells, CHO and some other cell types both without activation by upstream signaling and in active phosphorylated forms. GH treatment of cells stimulates phosphorylation of nuclear JAK2 without apparent changes of its subcellular localization. Active nuclear JAK2 phosphorylates transcription factor - nuclear factor 1-C2 (NF1-C2) in mammary epithelial cells[8,179,180]. Since both JAK1 and JAK2 are expressed constitutively in the nucleus and a number of cytokine receptors undergo nuclear translocation the realization of primary nuclear JAK-STAT signaling pathway seems very possible.
Primary nuclear STAT activation has also been observed. Nuclear ErbB1 has been shown recently to interact with STAT3 in the nucleus leading to transcriptional activation of inducible iNOS gene in breast carcinoma cells and this pathway may be associated with a high malignancy of other types of tumor cells with high level of nuclear ErbB1 and STAT3[10]. C-terminal 80-kDa fragment of ErbB4 with nuclear traffic activity may associate with STAT5a in the nucleus and transactivate gene expression[178,181]. Intracellular ErbB4 domain possesses a nuclear localization sequence essential for its nuclear accumulation. In cotransfection experiments nuclear translocation of intracellular ErbB4 domain has been shown to be required for nuclear translocation of STAT5a and ErbB4 function as a nuclear chaperone for STAT5a transcription factor as has been suggested[182].
Thus, primary nuclear JAK-STAT signaling may become important at certain stages of the cell cycle or be critical for some populations of rapidly dividing or neoplastic cells.
Is a direct nuclear JAK-STAT pathway related to cholangiocarcinoma development Intensive nuclear Prl receptor accumulation has been revealed in our laboratory in rat bile duct cells only after normal cholangiocyte transition to active proliferation during postnatal ontogenesis or after common bile duct ligation and is accompanied by elevation of total receptor content both in males and females[7]. Similarly, we have found nuclear localized Prl receptors with sharp elevation of their expression in samples of human cholangiocarcinoma tissues (Figure 3) as well as in liver transplanted rat cholangiocarcinoma cell line RS-1.
Figure 3.
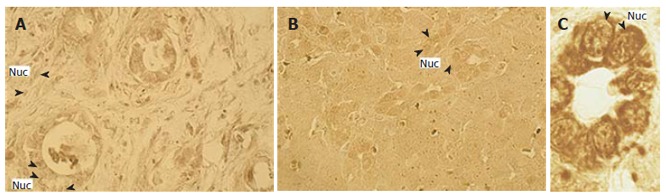
Immunohistochemistry for prolactin receptor (PrlR) expression in human cholangiocarcinoma sample (A) and in female (B) and male (C) rat liver tissue 14 d after common bile duct ligation. Intensive nuclear (Nuc), PrlR-posititive immunoreactivity is shown. Orig. mag, A, B: x 40; C: x 100.
Both total content and nuclear expression of Prl receptors in transplanted RS-1 rat cholangiocarcinoma cells show moderate sex dependence with female predominance (Table 4). The intrahepatic RS1 cholangiocarcinoma transplant induces nuclear accumulation of Prl receptors not only in malignant cholangiocytes but also in adjacent and distant hepatocytes and increases total Prl receptor immunoreactivity in these cells[7,58].
Table 4.
Nuclear prolactin receptor expression in rat liver transplanted RS-1 cholangiocarcinoma cells and adjacent hepatocytes as compared with normal cells
|
Nuclear PrlR-positive immunoreactivity (arbitrary units), medians (upper and lower quartiles) |
||||
| Normal cholangiocytes | RS-1 cholangiocarcinoma cells | Normal hepatocytes | Tumor adjacent hepatocytes | |
| Males | 47 (0; 104) | 191b (119; 288) | 40 (26; 90) | 308b (227; 381) |
| Females | 113 (47; 194) | 408b (256; 555) | 59 (41; 101) | 415b (266; 581) |
P < 0.001 as compared with correspondent intact group, Mann-Witney U-test.
We were unable to reveal prominent nuclear Prl receptor expression in hepatoma H27 cells after intrahepatic transplantation. Thus, nuclear accumulation of Prl receptors as well as their overexpression in intrahepatically transplanted RS1 cholangiocarcinoma cells and in hepatocytes of tumor bearing animals can serve as a specific marker of cholangiocarcinoma. Since we have used monoclonal antibodies U5 to the extracellular domain of rat Prl receptor, it was revealed that nuclear accumulation may be due to both full length Prl receptor or Prl binding protein as have been found for GH binding protein[9,172]. Appearance of Prl receptors in the nucleus is associated with Prl participation in the regulation of the proliferation process. This is confirmed by the data indicating that in Prl-dependent Nb2 lymphoma cells the cell cycle progression stops at the early G1 phase in the absence of Prl. Treatment of such cells with Prl leads to its transport to the nucleus followed by cell cycle progression[8,183].
In this respect unusual nuclear localization of prostaglandin E2 receptor EP1 that belongs to the G-protein coupled receptor superfamily detected in human cholangiocarcinoma cell lines seems very interesting[109].
Presented data show that primary nuclear JAK-STAT signaling may be relevant to cholangiocarcinoma progression and unusual nuclear localization of components of the JAK-STAT cascade may be considered as a predisposition to cholangiocarcinoma development.
CONCLUSION
While the role of GH, Prl and interleukins in normal cholangiocytes and pathophysiological states has been studied during the last decade, to date, the role of the JAK-STAT pathway in cholangiocarcinoma progression has not really been investigated and its importance is underestimated. Nevertheless, the summarized data provide a generalized view of the implication of the central cytokine signaling in tumor development of epithelial bile duct cells. A more detailed study of JAK-STAT involvement in cholangiocyte proliferative diseases can open the door for newer therapeutic strategies serving a real alternative to surgery.
Moreover, an engagement of the new model of cancer (cholangiocarcinoma) to studies of JAK-STAT signaling can give us an exhaustive understanding of its role in cancer development at different stages, which is not yet completely clarified (promotive role in neoplasia development[33] and inhibitory role in metastasis progression[45]). Moreover, it can help us to decipher new mechanisms of activation of the components of the JAK-STAT pathway like receptor and/or ligand nuclear localization.
ACKNOWLEDGMENTS
We thank Dr. Vera Ruda for critical reading of the manuscript.
Footnotes
S- Editor Liu Y L- Editor Rippe RA E- Editor Liu Y
References
- 1.Hebenstreit D, Horejs-Hoeck J, Duschl A. JAK/STAT-dependent gene regulation by cytokines. Drug News Perspect. 2005;18:243–249. doi: 10.1358/dnp.2005.18.4.908658. [DOI] [PubMed] [Google Scholar]
- 2.Sirica AE. Cholangiocarcinoma: molecular targeting strategies for chemoprevention and therapy. Hepatology. 2005;41:5–15. doi: 10.1002/hep.20537. [DOI] [PubMed] [Google Scholar]
- 3.Farazi PA, Zeisberg M, Glickman J, Zhang Y, Kalluri R, DePinho RA. Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Res. 2006;66:6622–6627. doi: 10.1158/0008-5472.CAN-05-4609. [DOI] [PubMed] [Google Scholar]
- 4.Marzioni M, Fava G, Benedetti A. Nervous and Neuroendocrine regulation of the pathophysiology of cholestasis and of biliary carcinogenesis. World J Gastroenterol. 2006;12:3471–3480. doi: 10.3748/wjg.v12.i22.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yokomuro S, Tsuji H, Lunz JG 3rd, Sakamoto T, Ezure T, Murase N, Demetris AJ. Growth control of human biliary epithelial cells by interleukin 6, hepatocyte growth factor, transforming growth factor beta1, and activin A: comparison of a cholangiocarcinoma cell line with primary cultures of non-neoplastic biliary epithelial cells. Hepatology. 2000;32:26–35. doi: 10.1053/jhep.2000.8535. [DOI] [PubMed] [Google Scholar]
- 6.Alvaro D, Mancino MG, Onori P, Franchitto A, Alpini G, Francis H, Glaser S, Gaudio E. Estrogens and the pathophysiology of the biliary tree. World J Gastroenterol. 2006;12:3537–3545. doi: 10.3748/wjg.v12.i22.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bogorad RL, Ostroukhova TY, Orlova AN, Rubtsov PM, Smirnova OV. Long isoform of prolactin receptor predominates in rat intrahepatic bile ducts and further increases under obstructive cholestasis. J Endocrinol. 2006;188:345–354. doi: 10.1677/joe.1.06468. [DOI] [PubMed] [Google Scholar]
- 8.Smirnova OV. A new level of hormonal signal transduction: primary nuclear action of protein and peptide hormones. Membr Cell Biol. 2000;13:245–261. [PubMed] [Google Scholar]
- 9.Mertani HC, Raccurt M, Abbate A, Kindblom J, Törnell J, Billestrup N, Usson Y, Morel G, Lobie PE. Nuclear translocation and retention of growth hormone. Endocrinology. 2003;144:3182–3195. doi: 10.1210/en.2002-221121. [DOI] [PubMed] [Google Scholar]
- 10.Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2006;94:184–188. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perret-Vivancos C, Abbate A, Ardail D, Raccurt M, Usson Y, Lobie PE, Morel G. Growth hormone activity in mitochondria depends on GH receptor Box 1 and involves caveolar pathway targeting. Exp Cell Res. 2006;312:215–232. doi: 10.1016/j.yexcr.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 12.Brown RJ, Adams JJ, Pelekanos RA, Wan Y, McKinstry WJ, Palethorpe K, Seeber RM, Monks TA, Eidne KA, Parker MW, et al. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat Struct Mol Biol. 2005;12:814–821. doi: 10.1038/nsmb977. [DOI] [PubMed] [Google Scholar]
- 13.Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19:2607–2611. doi: 10.1038/sj.onc.1203478. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Shaw PE. Autocrine-mediated activation of STAT3 correlates with cell proliferation in breast carcinoma lines. J Biol Chem. 2002;277:17397–17405. doi: 10.1074/jbc.M109962200. [DOI] [PubMed] [Google Scholar]
- 15.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol. 2005;2:92–100. [PubMed] [Google Scholar]
- 17.Valentino L, Pierre J. JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol. 2006;71:713–721. doi: 10.1016/j.bcp.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 18.Auernhammer CJ, Melmed S. The central role of SOCS-3 in integrating the neuro-immunoendocrine interface. J Clin Invest. 2001;108:1735–1740. doi: 10.1172/JCI14662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehmann U, Schmitz J, Weissenbach M, Sobota RM, Hortner M, Friederichs K, Behrmann I, Tsiaris W, Sasaki A, Schneider-Mergener J, et al. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J Biol Chem. 2003;278:661–671. doi: 10.1074/jbc.M210552200. [DOI] [PubMed] [Google Scholar]
- 20.Yang XP, Schaper F, Teubner A, Lammert F, Heinrich PC, Matern S, Siewert E. Interleukin-6 plays a crucial role in the hepatic expression of SOCS3 during acute inflammatory processes in vivo. J Hepatol. 2005;43:704–710. doi: 10.1016/j.jhep.2005.02.048. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell TJ, John S. Signal transducer and activator of transcription (STAT) signalling and T-cell lymphomas. Immunology. 2005;114:301–312. doi: 10.1111/j.1365-2567.2005.02091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boccaccio C, Andò M, Tamagnone L, Bardelli A, Michieli P, Battistini C, Comoglio PM. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature. 1998;391:285–288. doi: 10.1038/34657. [DOI] [PubMed] [Google Scholar]
- 23.Olayioye MA, Beuvink I, Horsch K, Daly JM, Hynes NE. ErbB receptor-induced activation of stat transcription factors is mediated by Src tyrosine kinases. J Biol Chem. 1999;274:17209–17218. doi: 10.1074/jbc.274.24.17209. [DOI] [PubMed] [Google Scholar]
- 24.Gouilleux-Gruart V, Debierre-Grockiego F, Gouilleux F, Capiod JC, Claisse JF, Delobel J, Prin L. Activated Stat related transcription factors in acute leukemia. Leuk Lymphoma. 1997;28:83–88. doi: 10.3109/10428199709058334. [DOI] [PubMed] [Google Scholar]
- 25.Gao B, Shen X, Kunos G, Meng Q, Goldberg ID, Rosen EM, Fan S. Constitutive activation of JAK-STAT3 signaling by BRCA1 in human prostate cancer cells. FEBS Lett. 2001;488:179–184. doi: 10.1016/s0014-5793(00)02430-3. [DOI] [PubMed] [Google Scholar]
- 26.Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA, Cox CE, Falcone R, Fairclough R, Parsons S, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20:2499–2513. doi: 10.1038/sj.onc.1204349. [DOI] [PubMed] [Google Scholar]
- 27.Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffé M, Berthou C, Lessard M, Berger R, Ghysdael J, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Turkson J, Carter-Su C, Smithgall T, Levitzki A, Kraker A, Krolewski JJ, Medveczky P, Jove R. Activation of Stat3 in v-Src-transformed fibroblasts requires cooperation of Jak1 kinase activity. J Biol Chem. 2000;275:24935–24944. doi: 10.1074/jbc.M002383200. [DOI] [PubMed] [Google Scholar]
- 29.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 30.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE Jr. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 31.Kelly JA, Spolski R, Kovanen PE, Suzuki T, Bollenbacher J, Pise-Masison CA, Radonovich MF, Lee S, Jenkins NA, Copeland NG, et al. Stat5 synergizes with T cell receptor/antigen stimulation in the development of lymphoblastic lymphoma. J Exp Med. 2003;198:79–89. doi: 10.1084/jem.20021548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 33.Kindblom J, Dillner K, Sahlin L, Robertson F, Ormandy C, Törnell J, Wennbo H. Prostate hyperplasia in a transgenic mouse with prostate-specific expression of prolactin. Endocrinology. 2003;144:2269–2278. doi: 10.1210/en.2002-0187. [DOI] [PubMed] [Google Scholar]
- 34.Manhes C, Goffin V, Kelly PA, Touraine P. Autocrine prolactin as a promotor of mammary tumour growth. J Dairy Res. 2005;72 Spec No:58–65. doi: 10.1017/s0022029905001196. [DOI] [PubMed] [Google Scholar]
- 35.Harvey PW, Everett DJ, Springall CJ. Hyperprolactinaemia as an adverse effect in regulatory and clinical toxicology: role in breast and prostate cancer. Hum Exp Toxicol. 2006;25:395–404. doi: 10.1191/0960327106ht643oa. [DOI] [PubMed] [Google Scholar]
- 36.Tworoger SS, Eliassen AH, Sluss P, Hankinson SE. A prospective study of plasma prolactin concentrations and risk of premenopausal and postmenopausal breast cancer. J Clin Oncol. 2007;25:1482–1488. doi: 10.1200/JCO.2006.07.6356. [DOI] [PubMed] [Google Scholar]
- 37.Törnell J, Carlsson B, Pohjanen P, Wennbo H, Rymo L, Isaksson O. High frequency of mammary adenocarcinomas in metallothionein promoter-human growth hormone transgenic mice created from two different strains of mice. J Steroid Biochem Mol Biol. 1992;43:237–242. doi: 10.1016/0960-0760(92)90213-3. [DOI] [PubMed] [Google Scholar]
- 38.Gutzman JH, Miller KK, Schuler LA. Endogenous human prolactin and not exogenous human prolactin induces estrogen receptor alpha and prolactin receptor expression and increases estrogen responsiveness in breast cancer cells. J Steroid Biochem Mol Biol. 2004;88:69–77. doi: 10.1016/j.jsbmb.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 39.Raccurt M, Lobie PE, Moudilou E, Garcia-Caballero T, Frappart L, Morel G, Mertani HC. High stromal and epithelial human gh gene expression is associated with proliferative disorders of the mammary gland. J Endocrinol. 2002;175:307–318. doi: 10.1677/joe.0.1750307. [DOI] [PubMed] [Google Scholar]
- 40.Mukhina S, Mertani HC, Guo K, Lee KO, Gluckman PD, Lobie PE. Phenotypic conversion of human mammary carcinoma cells by autocrine human growth hormone. Proc Natl Acad Sci USA. 2004;101:15166–15171. doi: 10.1073/pnas.0405881101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu T, Starling-Emerald B, Zhang X, Lee KO, Gluckman PD, Mertani HC, Lobie PE. Oncogenic transformation of human mammary epithelial cells by autocrine human growth hormone. Cancer Res. 2005;65:317–324. [PubMed] [Google Scholar]
- 42.Stoiber D, Kovacic B, Schuster C, Schellack C, Karaghiosoff M, Kreibich R, Weisz E, Artwohl M, Kleine OC, Muller M, et al. TYK2 is a key regulator of the surveillance of B lymphoid tumors. J Clin Invest. 2004;114:1650–1658. doi: 10.1172/JCI22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sexl V, Kovacic B, Piekorz R, Moriggl R, Stoiber D, Hoffmeyer A, Liebminger R, Kudlacek O, Weisz E, Rothammer K, et al. Jak1 deficiency leads to enhanced Abelson-induced B-cell tumor formation. Blood. 2003;101:4937–4943. doi: 10.1182/blood-2001-11-0142. [DOI] [PubMed] [Google Scholar]
- 44.Sultan AS, Xie J, LeBaron MJ, Ealley EL, Nevalainen MT, Rui H. Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene. 2005;24:746–760. doi: 10.1038/sj.onc.1208203. [DOI] [PubMed] [Google Scholar]
- 45.Nouhi Z, Chughtai N, Hartley S, Cocolakis E, Lebrun JJ, Ali S. Defining the role of prolactin as an invasion suppressor hormone in breast cancer cells. Cancer Res. 2006;66:1824–1832. doi: 10.1158/0008-5472.CAN-05-2292. [DOI] [PubMed] [Google Scholar]
- 46.Nevalainen MT, Xie J, Torhorst J, Bubendorf L, Haas P, Kononen J, Sauter G, Rui H. Signal transducer and activator of transcription-5 activation and breast cancer prognosis. J Clin Oncol. 2004;22:2053–2060. doi: 10.1200/JCO.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 47.Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr Rev. 1998;19:225–268. doi: 10.1210/edrv.19.3.0334. [DOI] [PubMed] [Google Scholar]
- 48.Horseman ND. Prolactin receptor diversity in humans: novel isoforms suggest general principles. Trends Endocrinol Metab. 2002;13:47–48. doi: 10.1016/s1043-2760(01)00558-6. [DOI] [PubMed] [Google Scholar]
- 49.Orlova AN, Smirnova OV, Turovetskiĭ VB, Smirnov AN. [Effect of bromocriptin on prolactin receptor expression in liver cells after the common bile duct ligation] Biull Eksp Biol Med. 1998;126:52–55. [PubMed] [Google Scholar]
- 50.Orlova AN, Smirnov AN, Smirnova OV. [The role of prolactin in the functional regulation of liver cells after the common bile duct ligation] Biull Eksp Biol Med. 1999;127:573–575. [PubMed] [Google Scholar]
- 51.Goffin V, Kelly PA. The prolactin/growth hormone receptor family: structure/function relationships. J Mammary Gland Biol Neoplasia. 1997;2:7–17. doi: 10.1023/a:1026313211704. [DOI] [PubMed] [Google Scholar]
- 52.Dif F, Saunier E, Demeneix B, Kelly PA, Edery M. Cytokine-inducible SH2-containing protein suppresses PRL signaling by binding the PRL receptor. Endocrinology. 2001;142:5286–5293. doi: 10.1210/endo.142.12.8549. [DOI] [PubMed] [Google Scholar]
- 53.Sutherland KD, Lindeman GJ, Visvader JE. Knocking off SOCS genes in the mammary gland. Cell Cycle. 2007;6:799–803. doi: 10.4161/cc.6.7.4037. [DOI] [PubMed] [Google Scholar]
- 54.Castagnetta LA, Agostara B, Montalto G, Polito L, Campisi I, Saetta A, Itoh T, Yu B, Chen S, Carruba G. Local estrogen formation by nontumoral, cirrhotic, and malignant human liver tissues and cells. Cancer Res. 2003;63:5041–5045. [PubMed] [Google Scholar]
- 55.Granata OM, Cocciadiferro L, Miceli V, Polito LM, Campisi I, Carruba G. Metabolic profiles of androgens in malignant human liver cell lines. Ann N Y Acad Sci. 2006;1089:262–267. doi: 10.1196/annals.1386.028. [DOI] [PubMed] [Google Scholar]
- 56.Simon-Holtorf J, Mönig H, Klomp HJ, Reinecke-Lüthge A, Fölsch UR, Kloehn S. Expression and distribution of prolactin receptor in normal, fibrotic, and cirrhotic human liver. Exp Clin Endocrinol Diabetes. 2006;114:584–589. doi: 10.1055/s-2006-948310. [DOI] [PubMed] [Google Scholar]
- 57.Zenkova TY, Kulikov AV, Bogorad RL, Rozenkrants AA, Platonova LV, Shono NI, Gal'perin EI, Smirnova OV. Expression of prolactin receptors in human liver during cholestasis of different etiology and secondary liver cancer. Bull Exp Biol Med. 2003;135:566–569. doi: 10.1023/a:1025429318932. [DOI] [PubMed] [Google Scholar]
- 58.Ostroukhova TY, Kulikov AV, Rozenkrants AA, Smirnova OV. Overexpression of prolactin receptors during intrahepatic transplantation of RS1 rat cholangiocellular carcinoma cells. Bull Exp Biol Med. 2006;141:364–367. doi: 10.1007/s10517-006-0172-6. [DOI] [PubMed] [Google Scholar]
- 59.Frasor J, Gibori G. Prolactin regulation of estrogen receptor expression. Trends Endocrinol Metab. 2003;14:118–123. doi: 10.1016/s1043-2760(03)00030-4. [DOI] [PubMed] [Google Scholar]
- 60.de Castillo B, Cawthorn S, Moppett J, Shere M, Norman M. Expression of prolactin receptor mRNA in oestrogen receptor positive breast cancers pre- and post-tamoxifen therapy. Eur J Surg Oncol. 2004;30:515–519. doi: 10.1016/j.ejso.2003.03.001. [DOI] [PubMed] [Google Scholar]
- 61.Leung KC, Johannsson G, Leong GM, Ho KK. Estrogen regulation of growth hormone action. Endocr Rev. 2004;25:693–721. doi: 10.1210/er.2003-0035. [DOI] [PubMed] [Google Scholar]
- 62.Dong J, Tsai-Morris CH, Dufau ML. A novel estradiol/estrogen receptor alpha-dependent transcriptional mechanism controls expression of the human prolactin receptor. J Biol Chem. 2006;281:18825–18836. doi: 10.1074/jbc.M512826200. [DOI] [PubMed] [Google Scholar]
- 63.Björnström L, Kilic E, Norman M, Parker MG, Sjöberg M. Cross-talk between Stat5b and estrogen receptor-alpha and -beta in mammary epithelial cells. J Mol Endocrinol. 2001;27:93–106. doi: 10.1677/jme.0.0270093. [DOI] [PubMed] [Google Scholar]
- 64.Faulds MH, Pettersson K, Gustafsson JA, Haldosén LA. Cross-talk between ERs and signal transducer and activator of transcription 5 is E2 dependent and involves two functionally separate mechanisms. Mol Endocrinol. 2001;15:1929–1940. doi: 10.1210/mend.15.11.0726. [DOI] [PubMed] [Google Scholar]
- 65.Cao J, Wood M, Liu Y, Hoffman T, Hyde J, Park-Sarge OK, Vore M. Estradiol represses prolactin-induced expression of Na+/taurocholate cotransporting polypeptide in liver cells through estrogen receptor-alpha and signal transducers and activators of transcription 5a. Endocrinology. 2004;145:1739–1749. doi: 10.1210/en.2003-0752. [DOI] [PubMed] [Google Scholar]
- 66.Wang Y, Cheng CH. ERalpha and STAT5a cross-talk: interaction through C-terminal portions of the proteins decreases STAT5a phosphorylation, nuclear translocation and DNA-binding. FEBS Lett. 2004;572:238–244. doi: 10.1016/j.febslet.2004.06.098. [DOI] [PubMed] [Google Scholar]
- 67.Alvaro D, Barbaro B, Franchitto A, Onori P, Glaser SS, Alpini G, Francis H, Marucci L, Sterpetti P, Ginanni-Corradini S, et al. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am J Pathol. 2006;169:877–888. doi: 10.2353/ajpath.2006.050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Acalovschi M. Cholangiocarcinoma: risk factors, diagnosis and management. Rom J Intern Med. 2004;42:41–58. [PubMed] [Google Scholar]
- 69.Huang Y, Li X, Jiang J, Frank SJ. Prolactin modulates phosphorylation, signaling and trafficking of epidermal growth factor receptor in human T47D breast cancer cells. Oncogene. 2006;25:7565–7576. doi: 10.1038/sj.onc.1209740. [DOI] [PubMed] [Google Scholar]
- 70.Flores-Morales A, Greenhalgh CJ, Norstedt G, Rico-Bautista E. Negative regulation of growth hormone receptor signaling. Mol Endocrinol. 2006;20:241–253. doi: 10.1210/me.2005-0170. [DOI] [PubMed] [Google Scholar]
- 71.Waters MJ, Hoang HN, Fairlie DP, Pelekanos RA, Brown RJ. New insights into growth hormone action. J Mol Endocrinol. 2006;36:1–7. doi: 10.1677/jme.1.01933. [DOI] [PubMed] [Google Scholar]
- 72.Alvaro D, Metalli VD, Alpini G, Onori P, Franchitto A, Barbaro B, Glaser SS, Francis H, Cantafora A, Blotta I, et al. The intrahepatic biliary epithelium is a target of the growth hormone/insulin-like growth factor 1 axis. J Hepatol. 2005;43:875–883. doi: 10.1016/j.jhep.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 73.Held MA, Cosme-Blanco W, Difedele LM, Bonkowski EL, Menon RK, Denson LA. Alterations in growth hormone receptor abundance regulate growth hormone signaling in murine obstructive cholestasis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G986–G993. doi: 10.1152/ajpgi.00287.2004. [DOI] [PubMed] [Google Scholar]
- 74.Dieudonné MN, Sammari A, Dos Santos E, Leneveu MC, Giudicelli Y, Pecquery R. Sex steroids and leptin regulate 11beta-hydroxysteroid dehydrogenase I and P450 aromatase expressions in human preadipocytes: Sex specificities. J Steroid Biochem Mol Biol. 2006;99:189–196. doi: 10.1016/j.jsbmb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 75.Frühbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393:7–20. doi: 10.1042/BJ20051578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sulkowska M, Golaszewska J, Wincewicz A, Koda M, Baltaziak M, Sulkowski S. Leptin--from regulation of fat metabolism to stimulation of breast cancer growth. Pathol Oncol Res. 2006;12:69–72. doi: 10.1007/BF02893446. [DOI] [PubMed] [Google Scholar]
- 77.Wake DJ, Strand M, Rask E, Westerbacka J, Livingstone DE, Soderberg S, Andrew R, Yki-Jarvinen H, Olsson T, Walker BR. Intra-adipose sex steroid metabolism and body fat distribution in idiopathic human obesity. Clin Endocrinol (Oxf) 2007;66:440–446. doi: 10.1111/j.1365-2265.2007.02755.x. [DOI] [PubMed] [Google Scholar]
- 78.Somasundar P, Frankenberry KA, Skinner H, Vedula G, McFadden DW, Riggs D, Jackson B, Vangilder R, Hileman SM, Vona-Davis LC. Prostate cancer cell proliferation is influenced by leptin. J Surg Res. 2004;118:71–82. doi: 10.1016/j.jss.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 79.Eguchi M, Gillis LC, Liu Y, Lyakhovsky N, Du M, McDermott JC, Sweeney G. Regulation of SOCS-3 expression by leptin and its co-localization with insulin receptor in rat skeletal muscle cells. Mol Cell Endocrinol. 2007;267:38–45. doi: 10.1016/j.mce.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 80.O'brien SN, Welter BH, Price TM. Presence of leptin in breast cell lines and breast tumors. Biochem Biophys Res Commun. 1999;259:695–698. doi: 10.1006/bbrc.1999.0843. [DOI] [PubMed] [Google Scholar]
- 81.Briscoe CP, Hanif S, Arch JR, Tadayyon M. Leptin receptor long-form signalling in a human liver cell line. Cytokine. 2001;14:225–229. doi: 10.1006/cyto.2001.0871. [DOI] [PubMed] [Google Scholar]
- 82.Saxena NK, Titus MA, Ding X, Floyd J, Srinivasan S, Sitaraman SV, Anania FA. Leptin as a novel profibrogenic cytokine in hepatic stellate cells: mitogenesis and inhibition of apoptosis mediated by extracellular regulated kinase (Erk) and Akt phosphorylation. FASEB J. 2004;18:1612–1614. doi: 10.1096/fj.04-1847fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ding X, Saxena NK, Lin S, Xu A, Srinivasan S, Anania FA. The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol. 2005;166:1655–1669. doi: 10.1016/S0002-9440(10)62476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bahçecioğlu IH, Yalniz M, Ataseven H, Bülbüller N, Keçeci M, Demirdağ K, Ozercan I, Ustündağ B. TNF-alpha and leptin in experimental liver fibrosis models induced by carbon tetrachloride and by common bile duct ligation. Cell Biochem Funct. 2004;22:359–363. doi: 10.1002/cbf.1114. [DOI] [PubMed] [Google Scholar]
- 85.Sayed-Ahmed A, Rudas P, Bartha T. Partial cloning and localisation of leptin and its receptor in the one-humped camel (Camelus dromedarius) Vet J. 2005;170:264–267. doi: 10.1016/j.tvjl.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 86.Sasaki R. Pleiotropic functions of erythropoietin. Intern Med. 2003;42:142–149. doi: 10.2169/internalmedicine.42.142. [DOI] [PubMed] [Google Scholar]
- 87.Udupa KB. Functional significance of erythropoietin receptor on tumor cells. World J Gastroenterol. 2006;12:7460–7462. doi: 10.3748/wjg.v12.i46.7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ohneda O, Yanai N, Obinata M. Erythropoietin as a mitogen for fetal liver stromal cells which support erythropoiesis. Exp Cell Res. 1993;208:327–331. doi: 10.1006/excr.1993.1253. [DOI] [PubMed] [Google Scholar]
- 89.Yasuda Y, Fujita Y, Matsuo T, Koinuma S, Hara S, Tazaki A, Onozaki M, Hashimoto M, Musha T, Ogawa K, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis. 2003;24:1021–1029. doi: 10.1093/carcin/bgg060. [DOI] [PubMed] [Google Scholar]
- 90.Hardee ME, Arcasoy MO, Blackwell KL, Kirkpatrick JP, Dewhirst MW. Erythropoietin biology in cancer. Clin Cancer Res. 2006;12:332–339. doi: 10.1158/1078-0432.CCR-05-1771. [DOI] [PubMed] [Google Scholar]
- 91.Naughton GK, Naughton BA, Gordon AS. Erythropoietin production by macrophages in the regenerating liver. J Surg Oncol. 1985;30:184–197. doi: 10.1002/jso.2930300312. [DOI] [PubMed] [Google Scholar]
- 92.Westphal G, Niederberger E, Blum C, Wollman Y, Knoch TA, Rebel W, Debus J, Friedrich E. Erythropoietin and G-CSF receptors in human tumor cells: expression and aspects regarding functionality. Tumori. 2002;88:150–159. doi: 10.1177/030089160208800214. [DOI] [PubMed] [Google Scholar]
- 93.Nakamatsu K, Nishimura Y, Suzuki M, Kanamori S, Maenishi O, Yasuda Y. Erythropoietin/erythropoietin-receptor system as an angiogenic factor in chemically induced murine hepatic tumors. Int J Clin Oncol. 2004;9:184–188. doi: 10.1007/s10147-004-0399-z. [DOI] [PubMed] [Google Scholar]
- 94.Cai YF, Zhen ZJ, Min J, Fang TL, Chu ZH, Chen JS. Selection, proliferation and differentiation of bone marrow-derived liver stem cells with a culture system containing cholestatic serum in vitro. World J Gastroenterol. 2004;10:3308–3312. doi: 10.3748/wjg.v10.i22.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alison MR, Lovell MJ. Liver cancer: the role of stem cells. Cell Prolif. 2005;38:407–421. doi: 10.1111/j.1365-2184.2005.00354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Libbrecht L. Hepatic progenitor cells in human liver tumor development. World J Gastroenterol. 2006;12:6261–6265. doi: 10.3748/wjg.v12.i39.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818–3822. doi: 10.1038/sj.onc.1209558. [DOI] [PubMed] [Google Scholar]
- 98.Fried W, Barone-Varelas J, Morley C. Factors that regulate extrarenal erythropoietin production. Blood Cells. 1984;10:287–304. [PubMed] [Google Scholar]
- 99.Yasuda Y, Masuda S, Chikuma M, Inoue K, Nagao M, Sasaki R. Estrogen-dependent production of erythropoietin in uterus and its implication in uterine angiogenesis. J Biol Chem. 1998;273:25381–25387. doi: 10.1074/jbc.273.39.25381. [DOI] [PubMed] [Google Scholar]
- 100.Masuda S, Kobayashi T, Chikuma M, Nagao M, Sasaki R. The oviduct produces erythropoietin in an estrogen- and oxygen-dependent manner. Am J Physiol Endocrinol Metab. 2000;278:E1038–E1044. doi: 10.1152/ajpendo.2000.278.6.E1038. [DOI] [PubMed] [Google Scholar]
- 101.Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–2556. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- 102.Ishikawa H, Tsuyama N, Obata M, M Kawano M. Mitogenic signals initiated via interleukin-6 receptor complexes in cooperation with other transmembrane molecules in myelomas. J Clin Exp Hematop. 2006;46:55–66. doi: 10.3960/jslrt.46.55. [DOI] [PubMed] [Google Scholar]
- 103.Lamireau T, Zoltowska M, Levy E, Yousef I, Rosenbaum J, Tuchweber B, Desmoulière A. Effects of bile acids on biliary epithelial cells: proliferation, cytotoxicity, and cytokine secretion. Life Sci. 2003;72:1401–1411. doi: 10.1016/s0024-3205(02)02408-6. [DOI] [PubMed] [Google Scholar]
- 104.Demetris AJ, Lunz JG 3rd, Specht S, Nozaki I. Biliary wound healing, ductular reactions, and IL-6/gp130 signaling in the development of liver disease. World J Gastroenterol. 2006;12:3512–3522. doi: 10.3748/wjg.v12.i22.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yokoyama T, Komori A, Nakamura M, Takii Y, Kamihira T, Shimoda S, Mori T, Fujiwara S, Koyabu M, Taniguchi K, et al. Human intrahepatic biliary epithelial cells function in innate immunity by producing IL-6 and IL-8 via the TLR4-NF-kappaB and -MAPK signaling pathways. Liver Int. 2006;26:467–476. doi: 10.1111/j.1478-3231.2006.01254.x. [DOI] [PubMed] [Google Scholar]
- 106.Tangkijvanich P, Thong-ngam D, Theamboonlers A, Hanvivatvong O, Kullavanijaya P, Poovorawan Y. Diagnostic role of serum interleukin 6 and CA 19-9 in patients with cholangiocarcinoma. Hepatogastroenterology. 2006;51:15–19. [PubMed] [Google Scholar]
- 107.Shimizu T, Yokomuro S, Mizuguchi Y, Kawahigashi Y, Arima Y, Taniai N, Mamada Y, Yoshida H, Akimaru K, Tajiri T. Effect of transforming growth factor-beta1 on human intrahepatic cholangiocarcinoma cell growth. World J Gastroenterol. 2006;12:6316–6324. doi: 10.3748/wjg.v12.i39.6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fernández-Martínez E, Pérez-Alvarez V, Tsutsumi V, Shibayama M, Muriel P. Chronic bile duct obstruction induces changes in plasma and hepatic levels of cytokines and nitric oxide in the rat. Exp Toxicol Pathol. 2006;58:49–58. doi: 10.1016/j.etp.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 109.Han C, Demetris AJ, Stolz DB, Xu L, Lim K, Wu T. Modulation of Stat3 activation by the cytosolic phospholipase A2alpha and cyclooxygenase-2-controlled prostaglandin E2 signaling pathway. J Biol Chem. 2006;281:24831–24846. doi: 10.1074/jbc.M602201200. [DOI] [PubMed] [Google Scholar]
- 110.Meng F, Yamagiwa Y, Ueno Y, Patel T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J Hepatol. 2006;44:1055–1065. doi: 10.1016/j.jhep.2005.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Isomoto H, Kobayashi S, Werneburg NW, Bronk SF, Guicciardi ME, Frank DA, Gores GJ. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005;42:1329–1338. doi: 10.1002/hep.20966. [DOI] [PubMed] [Google Scholar]
- 112.Isomoto H, Mott JL, Kobayashi S, Werneburg NW, Bronk SF, Haan S, Gores GJ. Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing. Gastroenterology. 2007;132:384–396. doi: 10.1053/j.gastro.2006.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hung W, Elliott B. Co-operative effect of c-Src tyrosine kinase and Stat3 in activation of hepatocyte growth factor expression in mammary carcinoma cells. J Biol Chem. 2001;276:12395–12403. doi: 10.1074/jbc.M010715200. [DOI] [PubMed] [Google Scholar]
- 114.Tomida M, Saito T. The human hepatocyte growth factor (HGF) gene is transcriptionally activated by leukemia inhibitory factor through the Stat binding element. Oncogene. 2004;23:679–686. doi: 10.1038/sj.onc.1207190. [DOI] [PubMed] [Google Scholar]
- 115.Devarajan P. Has HGF met other partners Met-independent epithelial morphogenesis induced by HGF. focus on "Hepatocyte growth factor induces MDCK cell morphogenesis without causing loss of tight junction functional integrity". Am J Physiol Cell Physiol. 2004;286:C475–C477. doi: 10.1152/ajpcell.00517.2003. [DOI] [PubMed] [Google Scholar]
- 116.Gentile A, Comoglio PM. Invasive growth: a genetic program. Int J Dev Biol. 2004;48:451–456. doi: 10.1387/ijdb.041799ag. [DOI] [PubMed] [Google Scholar]
- 117.Bolanos-Garcia VM. MET meet adaptors: functional and structural implications in downstream signalling mediated by the Met receptor. Mol Cell Biochem. 2005;276:149–157. doi: 10.1007/s11010-005-3696-6. [DOI] [PubMed] [Google Scholar]
- 118.Runge DM, Runge D, Foth H, Strom SC, Michalopoulos GK. STAT 1alpha/1beta, STAT 3 and STAT 5: expression and association with c-MET and EGF-receptor in long-term cultures of human hepatocytes. Biochem Biophys Res Commun. 1999;265:376–381. doi: 10.1006/bbrc.1999.1681. [DOI] [PubMed] [Google Scholar]
- 119.Gahr S, Merger M, Bollheimer LC, Hammerschmied CG, Schölmerich J, Hügl SR. Hepatocyte growth factor stimulates proliferation of pancreatic beta-cells particularly in the presence of subphysiological glucose concentrations. J Mol Endocrinol. 2002;28:99–110. doi: 10.1677/jme.0.0280099. [DOI] [PubMed] [Google Scholar]
- 120.Tokumaru S, Sayama K, Yamasaki K, Shirakata Y, Hanakawa Y, Yahata Y, Dai X, Tohyama M, Yang L, Yoshimura A, et al. SOCS3/CIS3 negative regulation of STAT3 in HGF-induced keratinocyte migration. Biochem Biophys Res Commun. 2005;327:100–105. doi: 10.1016/j.bbrc.2004.11.145. [DOI] [PubMed] [Google Scholar]
- 121.Wang D, Li Z, Messing EM, Wu G. The SPRY domain-containing SOCS box protein 1 (SSB-1) interacts with MET and enhances the hepatocyte growth factor-induced Erk-Elk-1-serum response element pathway. J Biol Chem. 2005;280:16393–16401. doi: 10.1074/jbc.M413897200. [DOI] [PubMed] [Google Scholar]
- 122.Masters SL, Yao S, Willson TA, Zhang JG, Palmer KR, Smith BJ, Babon JJ, Nicola NA, Norton RS, Nicholson SE. The SPRY domain of SSB-2 adopts a novel fold that presents conserved Par-4-binding residues. Nat Struct Mol Biol. 2006;13:77–84. doi: 10.1038/nsmb1034. [DOI] [PubMed] [Google Scholar]
- 123.Endo K, Yoon BI, Pairojkul C, Demetris AJ, Sirica AE. ERBB-2 overexpression and cyclooxygenase-2 up-regulation in human cholangiocarcinoma and risk conditions. Hepatology. 2002;36:439–450. doi: 10.1053/jhep.2002.34435. [DOI] [PubMed] [Google Scholar]
- 124.Li Z, Mizuno S, Nakamura T. Antinecrotic and antiapoptotic effects of hepatocyte growth factor on cholestatic hepatitis in a mouse model of bile-obstructive diseases. Am J Physiol Gastrointest Liver Physiol. 2007;292:G639–G646. doi: 10.1152/ajpgi.00292.2006. [DOI] [PubMed] [Google Scholar]
- 125.Purps O, Lahme B, Gressner AM, Meindl-Beinker NM, Dooley S. Loss of TGF-beta dependent growth control during HSC transdifferentiation. Biochem Biophys Res Commun. 2007;353:841–847. doi: 10.1016/j.bbrc.2006.12.125. [DOI] [PubMed] [Google Scholar]
- 126.Wu F, Wu L, Zheng S, Ding W, Teng L, Wang Z, Ma Z, Zhao W. The clinical value of hepatocyte growth factor and its receptor--c-met for liver cancer patients with hepatectomy. Dig Liver Dis. 2006;38:490–497. doi: 10.1016/j.dld.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 127.Imai Y, Kubota Y, Yamamoto S, Tsuji K, Shimatani M, Shibatani N, Takamido S, Matsushita M, Okazaki K. Neutrophils enhance invasion activity of human cholangiocellular carcinoma and hepatocellular carcinoma cells: an in vitro study. J Gastroenterol Hepatol. 2005;20:287–293. doi: 10.1111/j.1440-1746.2004.03575.x. [DOI] [PubMed] [Google Scholar]
- 128.Sasaki M, Ikeda H, Kataoka H, Nakanuma Y. Augmented expression of hepatocytes growth factor activator inhibitor type 1 (HAI-1) in intrahepatic small bile ducts in primary biliary cirrhosis. Virchows Arch. 2006;449:462–471. doi: 10.1007/s00428-006-0257-7. [DOI] [PubMed] [Google Scholar]
- 129.Radaeva S, Ferreira-Gonzalez A, Sirica AE. Overexpression of C-NEU and C-MET during rat liver cholangiocarcinogenesis: A link between biliary intestinal metaplasia and mucin-producing cholangiocarcinoma. Hepatology. 1999;29:1453–1462. doi: 10.1002/hep.510290524. [DOI] [PubMed] [Google Scholar]
- 130.Nakazawa K, Dobashi Y, Suzuki S, Fujii H, Takeda Y, Ooi A. Amplification and overexpression of c-erbB-2, epidermal growth factor receptor, and c-met in biliary tract cancers. J Pathol. 2005;206:356–365. doi: 10.1002/path.1779. [DOI] [PubMed] [Google Scholar]
- 131.Leelawat K, Leelawat S, Tepaksorn P, Rattanasinganchan P, Leungchaweng A, Tohtong R, Sobhon P. Involvement of c-Met/hepatocyte growth factor pathway in cholangiocarcinoma cell invasion and its therapeutic inhibition with small interfering RNA specific for c-Met. J Surg Res. 2006;136:78–84. doi: 10.1016/j.jss.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 132.DeArmond D, Brattain MG, Jessup JM, Kreisberg J, Malik S, Zhao S, Freeman JW. Autocrine-mediated ErbB-2 kinase activation of STAT3 is required for growth factor independence of pancreatic cancer cell lines. Oncogene. 2003;22:7781–7795. doi: 10.1038/sj.onc.1206966. [DOI] [PubMed] [Google Scholar]
- 133.Nautiyal J, Rishi AK, Majumdar AP. Emerging therapies in gastrointestinal cancers. World J Gastroenterol. 2006;12:7440–7450. doi: 10.3748/wjg.v12.i46.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cacalano NA, Sanden D, Johnston JA. Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat Cell Biol. 2001;3:460–465. doi: 10.1038/35074525. [DOI] [PubMed] [Google Scholar]
- 135.Kario E, Marmor MD, Adamsky K, Citri A, Amit I, Amariglio N, Rechavi G, Yarden Y. Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J Biol Chem. 2005;280:7038–7048. doi: 10.1074/jbc.M408575200. [DOI] [PubMed] [Google Scholar]
- 136.Nicholson SE, Metcalf D, Sprigg NS, Columbus R, Walker F, Silva A, Cary D, Willson TA, Zhang JG, Hilton DJ, et al. Suppressor of cytokine signaling (SOCS)-5 is a potential negative regulator of epidermal growth factor signaling. Proc Natl Acad Sci USA. 2005;102:2328–2333. doi: 10.1073/pnas.0409675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cohenuram M, Saif MW. Epidermal growth factor receptor inhibition strategies in pancreatic cancer: past, present and the future. JOP. 2007;8:4–15. [PubMed] [Google Scholar]
- 138.Terada T, Ashida K, Endo K, Horie S, Maeta H, Matsunaga Y, Takashima K, Ohta T, Kitamura Y. c-erbB-2 protein is expressed in hepatolithiasis and cholangiocarcinoma. Histopathology. 1998;33:325–331. doi: 10.1046/j.1365-2559.1998.00496.x. [DOI] [PubMed] [Google Scholar]
- 139.Ukita Y, Kato M, Terada T. Gene amplification and mRNA and protein overexpression of c-erbB-2 (HER-2/neu) in human intrahepatic cholangiocarcinoma as detected by fluorescence in situ hybridization, in situ hybridization, and immunohistochemistry. J Hepatol. 2002;36:780–785. doi: 10.1016/s0168-8278(02)00057-0. [DOI] [PubMed] [Google Scholar]
- 140.Altimari A, Fiorentino M, Gabusi E, Gruppioni E, Corti B, D'Errico A, Grigioni WF. Investigation of ErbB1 and ErbB2 expression for therapeutic targeting in primary liver tumours. Dig Liver Dis. 2003;35:332–338. doi: 10.1016/s1590-8658(03)00077-x. [DOI] [PubMed] [Google Scholar]
- 141.Polimeno L, Azzarone A, Zeng QH, Panella C, Subbotin V, Carr B, Bouzahzah B, Francavilla A, Starzl TE. Cell proliferation and oncogene expression after bile duct ligation in the rat: evidence of a specific growth effect on bile duct cells. Hepatology. 1995;21:1070–1078. [PMC free article] [PubMed] [Google Scholar]
- 142.Su WC, Shiesh SC, Liu HS, Chen CY, Chow NH, Lin XZ. Expression of oncogene products HER2/Neu and Ras and fibrosis-related growth factors bFGF, TGF-beta, and PDGF in bile from biliary malignancies and inflammatory disorders. Dig Dis Sci. 2001;46:1387–1392. doi: 10.1023/a:1010619316436. [DOI] [PubMed] [Google Scholar]
- 143.Yeh CN, Maitra A, Lee KF, Jan YY, Chen MF. Thioacetamide-induced intestinal-type cholangiocarcinoma in rat: an animal model recapitulating the multi-stage progression of human cholangiocarcinoma. Carcinogenesis. 2004;25:631–636. doi: 10.1093/carcin/bgh037. [DOI] [PubMed] [Google Scholar]
- 144.Wiedmann M, Feisthammel J, Blüthner T, Tannapfel A, Kamenz T, Kluge A, Mössner J, Caca K. Novel targeted approaches to treating biliary tract cancer: the dual epidermal growth factor receptor and ErbB-2 tyrosine kinase inhibitor NVP-AEE788 is more efficient than the epidermal growth factor receptor inhibitors gefitinib and erlotinib. Anticancer Drugs. 2006;17:783–795. doi: 10.1097/01.cad.0000217433.48870.37. [DOI] [PubMed] [Google Scholar]
- 145.Chen WY, White ME, Wagner TE, Kopchick JJ. Functional antagonism between endogenous mouse growth hormone (GH) and a GH analog results in dwarf transgenic mice. Endocrinology. 1991;129:1402–1408. doi: 10.1210/endo-129-3-1402. [DOI] [PubMed] [Google Scholar]
- 146.Bernichtein S, Kayser C, Dillner K, Moulin S, Kopchick JJ, Martial JA, Norstedt G, Isaksson O, Kelly PA, Goffin V. Development of pure prolactin receptor antagonists. J Biol Chem. 2003;278:35988–35999. doi: 10.1074/jbc.M305687200. [DOI] [PubMed] [Google Scholar]
- 147.Kirchhofer D, Lipari MT, Santell L, Billeci KL, Maun HR, Sandoval WN, Moran P, Ridgway J, Eigenbrot C, Lazarus RA. Utilizing the activation mechanism of serine proteases to engineer hepatocyte growth factor into a Met antagonist. Proc Natl Acad Sci USA. 2007;104:5306–5311. doi: 10.1073/pnas.0700184104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Smirnova OV, Petrashchuk OM, Smirnov AN. [Induction of expression of prolactin receptors in cholangiocytes of male and female rats after ligation of the common bile duct] Biull Eksp Biol Med. 1998;125:66–70. [PubMed] [Google Scholar]
- 149.Liu J, Kern JA. Neuregulin-1 activates the JAK-STAT pathway and regulates lung epithelial cell proliferation. Am J Respir Cell Mol Biol. 2002;27:306–313. doi: 10.1165/rcmb.4850. [DOI] [PubMed] [Google Scholar]
- 150.Damera G, Xia B, Sachdev GP. IL-4 induced MUC4 enhancement in respiratory epithelial cells in vitro is mediated through JAK-3 selective signaling. Respir Res. 2006;7:39. doi: 10.1186/1465-9921-7-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hayakawa F, Naoe T. SFK-STAT pathway: an alternative and important way to malignancies. Ann N Y Acad Sci. 2006;1086:213–222. doi: 10.1196/annals.1377.002. [DOI] [PubMed] [Google Scholar]
- 152.Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55:237–245. doi: 10.1007/s00262-005-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, Kitagawa T, Yamamoto J, Kubo T, Yoshikawa H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene. 2005;24:6406–6417. doi: 10.1038/sj.onc.1208788. [DOI] [PubMed] [Google Scholar]
- 154.Tischoff I, Wittekind C, Tannapfel A. Role of epigenetic alterations in cholangiocarcinoma. J Hepatobiliary Pancreat Surg. 2006;13:274–279. doi: 10.1007/s00534-005-1055-3. [DOI] [PubMed] [Google Scholar]
- 155.Ward AC, Touw I, Yoshimura A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood. 2000;95:19–29. [PubMed] [Google Scholar]
- 156.Kochendoerfer SK, Krishnan N, Buckley DJ, Buckley AR. Prolactin regulation of Bcl-2 family members: increased expression of bcl-xL but not mcl-1 or bad in Nb2-T cells. J Endocrinol. 2003;178:265–273. doi: 10.1677/joe.0.1780265. [DOI] [PubMed] [Google Scholar]
- 157.Xu Q, Briggs J, Park S, Niu G, Kortylewski M, Zhang S, Gritsko T, Turkson J, Kay H, Semenza GL, et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene. 2005;24:5552–5560. doi: 10.1038/sj.onc.1208719. [DOI] [PubMed] [Google Scholar]
- 158.Koon HW, Zhao D, Zhan Y, Rhee SH, Moyer MP, Pothoulakis C. Substance P stimulates cyclooxygenase-2 and prostaglandin E2 expression through JAK-STAT activation in human colonic epithelial cells. J Immunol. 2006;176:5050–5059. doi: 10.4049/jimmunol.176.8.5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Townsend PA, Scarabelli TM, Davidson SM, Knight RA, Latchman DS, Stephanou A. STAT-1 interacts with p53 to enhance DNA damage-induced apoptosis. J Biol Chem. 2004;279:5811–5820. doi: 10.1074/jbc.M302637200. [DOI] [PubMed] [Google Scholar]
- 160.Flørenes VA, Lu C, Bhattacharya N, Rak J, Sheehan C, Slingerland JM, Kerbel RS. Interleukin-6 dependent induction of the cyclin dependent kinase inhibitor p21WAF1/CIP1 is lost during progression of human malignant melanoma. Oncogene. 1999;18:1023–1032. doi: 10.1038/sj.onc.1202382. [DOI] [PubMed] [Google Scholar]
- 161.Takahashi S, Harigae H, Kaku M, Sasaki T, Licht JD. Flt3 mutation activates p21WAF1/CIP1 gene expression through the action of STAT5. Biochem Biophys Res Commun. 2004;316:85–92. doi: 10.1016/j.bbrc.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 162.de Koning JP, Soede-Bobok AA, Ward AC, Schelen AM, Antonissen C, van Leeuwen D, Löwenberg B, Touw IP. STAT3-mediated differentiation and survival and of myeloid cells in response to granulocyte colony-stimulating factor: role for the cyclin-dependent kinase inhibitor p27(Kip1) Oncogene. 2000;19:3290–3298. doi: 10.1038/sj.onc.1203627. [DOI] [PubMed] [Google Scholar]
- 163.Sasaki R, Masuda S, Nagao M. Erythropoietin: multiple physiological functions and regulation of biosynthesis. Biosci Biotechnol Biochem. 2000;64:1775–1793. doi: 10.1271/bbb.64.1775. [DOI] [PubMed] [Google Scholar]
- 164.Gaemers IC, Vos HL, Volders HH, van der Valk SW, Hilkens J. A stat-responsive element in the promoter of the episialin/MUC1 gene is involved in its overexpression in carcinoma cells. J Biol Chem. 2001;276:6191–6199. doi: 10.1074/jbc.M009449200. [DOI] [PubMed] [Google Scholar]
- 165.Perrais M, Pigny P, Ducourouble MP, Petitprez D, Porchet N, Aubert JP, Van Seuningen I. Characterization of human mucin gene MUC4 promoter: importance of growth factors and proinflammatory cytokines for its regulation in pancreatic cancer cells. J Biol Chem. 2001;276:30923–30933. doi: 10.1074/jbc.M104204200. [DOI] [PubMed] [Google Scholar]
- 166.Grasl-Kraupp B, Schausberger E, Hufnagl K, Gerner C, Löw-Baselli A, Rossmanith W, Parzefall W, Schulte-Hermann R. A novel mechanism for mitogenic signaling via pro-transforming growth factor alpha within hepatocyte nuclei. Hepatology. 2002;35:1372–1380. doi: 10.1053/jhep.2002.33203. [DOI] [PubMed] [Google Scholar]
- 167.Ur E, Wilkinson DA, Morash BA, Wilkinson M. Leptin immunoreactivity is localized to neurons in rat brain. Neuroendocrinology. 2002;75:264–272. doi: 10.1159/000054718. [DOI] [PubMed] [Google Scholar]
- 168.Lo HW, Xia W, Wei Y, Ali-Seyed M, Huang SF, Hung MC. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res. 2005;65:338–348. [PubMed] [Google Scholar]
- 169.Lo HW, Hsu SC, Hung MC. EGFR signaling pathway in breast cancers: from traditional signal transduction to direct nuclear translocalization. Breast Cancer Res Treat. 2006;95:211–218. doi: 10.1007/s10549-005-9011-0. [DOI] [PubMed] [Google Scholar]
- 170.Rycyzyn MA, Clevenger CV. The intranuclear prolactin/cyclophilin B complex as a transcriptional inducer. Proc Natl Acad Sci USA. 2002;99:6790–6795. doi: 10.1073/pnas.092160699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Clevenger CV. Nuclear localization and function of polypeptide ligands and their receptors: a new paradigm for hormone specificity within the mammary gland. Breast Cancer Res. 2003;5:181–187. doi: 10.1186/bcr601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Graichen R, Sandstedt J, Goh EL, Isaksson OG, Törnell J, Lobie PE. The growth hormone-binding protein is a location-dependent cytokine receptor transcriptional enhancer. J Biol Chem. 2003;278:6346–6354. doi: 10.1074/jbc.M207546200. [DOI] [PubMed] [Google Scholar]
- 173.Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z, Ali-Seyed M, Lee DF, Bartholomeusz G, Ou-Yang F, et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell. 2004;6:251–261. doi: 10.1016/j.ccr.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 174.Giri DK, Ali-Seyed M, Li LY, Lee DF, Ling P, Bartholomeusz G, Wang SC, Hung MC. Endosomal transport of ErbB-2: mechanism for nuclear entry of the cell surface receptor. Mol Cell Biol. 2005;25:11005–11018. doi: 10.1128/MCB.25.24.11005-11018.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Psyrri A, Yu Z, Weinberger PM, Sasaki C, Haffty B, Camp R, Rimm D, Burtness BA. Quantitative determination of nuclear and cytoplasmic epidermal growth factor receptor expression in oropharyngeal squamous cell cancer by using automated quantitative analysis. Clin Cancer Res. 2005;11:5856–5862. doi: 10.1158/1078-0432.CCR-05-0420. [DOI] [PubMed] [Google Scholar]
- 176.Lo HW, Ali-Seyed M, Wu Y, Bartholomeusz G, Hsu SC, Hung MC. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J Cell Biochem. 2006;98:1570–1583. doi: 10.1002/jcb.20876. [DOI] [PubMed] [Google Scholar]
- 177.Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung MC. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 178.Muraoka-Cook RS, Sandahl M, Husted C, Hunter D, Miraglia L, Feng SM, Elenius K, Earp HS 3rd. The intracellular domain of ErbB4 induces differentiation of mammary epithelial cells. Mol Biol Cell. 2006;17:4118–4129. doi: 10.1091/mbc.E06-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lobie PE, Ronsin B, Silvennoinen O, Haldosén LA, Norstedt G, Morel G. Constitutive nuclear localization of Janus kinases 1 and 2. Endocrinology. 1996;137:4037–4045. doi: 10.1210/endo.137.9.8756581. [DOI] [PubMed] [Google Scholar]
- 180.Nilsson J, Bjursell G, Kannius-Janson M. Nuclear Jak2 and transcription factor NF1-C2: a novel mechanism of prolactin signaling in mammary epithelial cells. Mol Cell Biol. 2006;26:5663–5674. doi: 10.1128/MCB.02095-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Vidal GA, Naresh A, Marrero L, Jones FE. Presenilin-dependent gamma-secretase processing regulates multiple ERBB4/HER4 activities. J Biol Chem. 2005;280:19777–19783. doi: 10.1074/jbc.M412457200. [DOI] [PubMed] [Google Scholar]
- 182.Williams CC, Allison JG, Vidal GA, Burow ME, Beckman BS, Marrero L, Jones FE. The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J Cell Biol. 2004;167:469–478. doi: 10.1083/jcb.200403155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Buckley AR, Buckley DJ, Gout PW, Liang H, Rao YP, Blake MJ. Inhibition by genistein of prolactin-induced Nb2 lymphoma cell mitogenesis. Mol Cell Endocrinol. 1993;98:17–25. doi: 10.1016/0303-7207(93)90231-8. [DOI] [PubMed] [Google Scholar]