

Abstract
AIM: Heme oxygenase (HO)-1 catalyzes the conversion of heme to biliverdin, iron and carbon monoxide. HO-1 is induced by many stimuli including heme, Hb, heat stress, lipopolysaccharide (LPS) and cytokines. Previous studies demonstrated that LPS induced HO-1 gene activation and HO-1 expression in liver. However, the mechanisms of LPS-induced HO-1 expression in liver remain unknown. The effect of toll-like receptor-4 (TLR4) on LPS-induced liver HO-1 expression and the role of TNF-α and IL-1β in this condition were determined.
METHODS: HO- 1 expression was determined by immunofluorescent staining and immunoblotting. Double immunofluorescent staining was performed to determine the cell type of HO-1 expression in liver.
RESULTS: A low dose of LPS significantly increased HO-1 expression in the liver which was localized in Kupffer cells only. Furthermore, HO-1 expression was enhanced by three doses of LPS. HO-1 expression was significantly inhibited in the liver of TLR4 mutant mice. While the liver HO-1 expression in TNF KO mice was much lower than that in C57 mice following the same LPS treatment, IL-1β KO had a slight influence on liver HO-1 expression following LPS treatment.
CONCLUSION: The present results confirm that macrophages are the major source of HO-1 in the liver induced by LPS. This study demonstrates that TLR4 plays a dominant role in mediating HO-1 expression following LPS. LPS-induced HO-1 expression is mainly mediated by endogenous TNF-α, but only partially by endogenous IL-1β.
INTRODUCTION
Heme oxygenase (HO) catalyzes the degradation of heme into biliverdin, iron and carbon monoxide[1-3]. Heme oxygenase has three known isoforms that consist of the inducible isoform heme oxygenase-1 (HO-1), the constitutive isoform heme oxygenase-2 and heme oxygenase-3. HO-1 is induced not only by its substrate, hemin, but also by oxidative stress, heat stress, UV irradiation, heavy metals, lipopolysaccharide (LPS) and cytokines[4-8]. This diversity of HO-1 inducers has provided further support for the speculation that HO-1 may play a vital function in maintaining cellular homeostasis, and some studies suggest that HO-1 can serve as a key biological molecule in the adaptation and/or defense against oxidative stress.
LPS is generally regarded as a key initiating factor in the pathogenesis of septic shock, and it stimulates inflammatory cells to produce cytokines, and other inflammatory mediators. HO-1 is induced by LPS in some cultured cell models in vitro[4,9], and also in lungs[10], kidneys and spleen [11,12] in vivo. It has been reported that HO-1 is induced in the liver after administration of LPS[11,13,14], but little is known about the mechanism of LPS-induced HO-1 expression in liver.
Studies have demonstrated that LPS induces cellular response by the signaling molecules belonging to the Toll-like receptor (TLR) family[15]. So far, six members (TLR1-6) have been reported and two of these, TLR2 and TLR4, have been shown to be essential for the recognition of distinct bacterial cell wall components[15,16]. TLR4 recognizes LPS, lipoteichoic acid and Taxol[17-19]. Recently, it was demonstrated that TLR4 mediated heat shock protein 60 signaling[20]. Although TLR4 is clearly critical to LPS signaling, the role of TLR4 in LPS-induced HO-1 expression has remained unclear. TLR4 mutation attenuates LPS-induced NF-κB activation and the production of cytokines[21,22]. Previous studies showed that cytokines, TNF-α, IL-α, or IL-1β induced HO-1 expression in vitro[8,23,24]. Do endogenous cytokines, TNF-α and IL-1β play a role in LPS-induced HO-1 expression
The purposes of this study were to examine: the time course and localization of HO-1 expression in the liver by LPS stimulation, the role of TLR4 in LPS-induced liver HO-1 expression, whether LPS-induced HO-1 expression is dependent on TNF-α, and whether IL-1β plays a role in LPS-induced HO-1 expression.
MATERIALS AND METHODS
Animals
Male mice of the C57 BL/6, BALB/cJ and C.C3H-Tlr4Lps-d (TLR4 mutant) aged 8-10 weeks were obtained from Jackson Laboratory (Bar Harbor, ME). TNF-α knockout (TNF KO) mice of the same age range were generous gifts from Dr. David Riches of National Jewish Medical and Research Center (Denver, CO). IL-1β knockout (IL-1KO) mice were obtained from National Jewish Medical and Research Center (Denver, CO). The mice were kept on a 12-h light/dark cycle with free access to food and water. All animal experiments were approved by the University of Colorado Health Science Center Animal Care and Research Committee. During experiments, all the animals received humane care in compliance with the “Guide for the Care and Use of Laboratory Animals” [DHEW Publication No. (NIH) 85-23, revised in 1985, Office of Science and Health Reports, DRR/NIH, Bethesda, MD 20205].
Chemicals and reagents
Rabbit polyclonal antibody to HO-1 and the recombinant rat HO-1 were purchased from StressGen Biotechnologies Corp (Victoria, BC, Canada). Rat monoclonal antibody to mouse CD 68 was purchased from Serotec (Oxford, UK). Rat IgG and Cy3-conjugated goat anti-rabbit IgG were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Fluorescein-conjugated wheat germ agglutinin was obtained from Molecular Probes (Eugene, OR). LPS (Escherichia coli, O55:B5) and all other chemicals were obtained from Sigma (St Louis, MO).
Experimental protocols
To determine HO-1 expression in the liver, the mice received either LPS (0.5 mg/kg) or normal saline by tail vein injection. To determine a time course of HO-1 expression, a single dose, two or three doses of LPS were used in each every 24 hours. After anesthesia and heparinization (40 mg/kg of pentobarbital sodium, ip), the liver tissue samples were prepared for the assessment of HO-1 expression. A portion of the liver tissue was embedded in a tissue-freezing medium and frozen in dry-ice-chilled isopentane. The remaining liver tissue was frozen in liquid nitrogen. All the samples were stored at -70 °C prior to use.
Immunofluorescent detection of HO-1 expression
Liver HO-1 expression was determined by immunofluorescent staining as previously described[25]. Tissue cryosections (5 μm thick) were prepared with a cryostat (IEC Minotome plus, Needham Heights, MA) and collected on poly-l-lysine-coated slides. All incubations were performed at room temperature. Sections were treated with a mixture of 70% acetone and 30% methanol for 5 min, and then fixed with 3% paraformaldehyde for 20 min. Sections were washed with PBS, blocked with 10% normal goat serum for 30 min, and incubated for 1 h with a polyclonal rabbit anti-HO-1 antibody (1:300 in PBS containing 1% bovine serum albumin). Control sections were incubated with nonimmune rat IgG (5 mg/ml). After washed with PBS, sections were incubated with Cy3-conjugated goat anti-rabbit IgG (1:300 dilution with PBS containing 1% bovine serum albumin). The cell surface was counterstained with fluorescein-conjugated wheat germ agglutinin, and the nucleus with bis-benzimide. The sections were mounted with aqueous media. Microscopic analysis was performed with a Leica DMRXA digital microscope (Germany) equipped with Slidebook software (I. I. I. Inc., Denver, CO).
Double immunofluorescent staining of HO-1 and macrophage
Double immunofluorescent staining was performed to determine the cell type stained positively of HO-1. CD68 (macrophages)-positive cells were studied. Tissue cryosections (5-μm thick) were prepared, fixed as described for staining of HO-1. Sections were blocked with 5% normal goat serum + 5% normal donkey serum for 30 min. Then slides were incubated for 1 h with a rat monoclonal antibody to mouse macrophages (CD68, 1:200) and rabbit polyclonal antibody to HO-1 (1:300). Control sections were incubated with non-immune rat IgG and rabbit IgG. After washed with PBS, the sections were incubated with Cy3-conjugated donkey anti-rat IgG (CD68) and FITC-conjugated goat anti-rabbit IgG (HO-1). The nucleus was counterstained with bis-benzimiade. The sections were washed with PBS, and then mounted on aqueous mounting media. Microscopic observation and photography were performed as described above.
Immunoblotting of HO-1
Liver tissue was homogenized with 4 volumes of homogenate buffer (PBS containing the protease inhibitor cocktail and 1% Triton X100, pH 7.4), and centrifuged at 10000×g for 20 min at 4 °C. The resulting supernatant was collected and stored at -70 °C for immunoblotting of HO-1. Aliquots of protein (60 μg/lane) in the liver tissue homogenate were separated on a 4%-20% SDS-polyacrylamide gel (Bio-Rad, Hercules, CA). Separated proteins were blotted onto a nitrocellulose membrane (Bio-Rad, Hercules, CA), and equal loading was confirmed by densitometric assessment of protein band staining with Ponceau S solution. The membrane was rinsed in PBS and blocked for 1 h at room temperature with 5% dry milk in PBS. The membrane was then incubated with a polyclonal rabbit anti-HO-1 antibody (1:1000 dilution with 5% dry milk in PBST) at room temperature for 1 h. Following an incubation with HRP-conjugated goat anti-rabbit IgG (1:10000 dilution with 5% dry milk in PBST), HO-1 bands were developed using ECL and visualized by exposure to Kodak X-Omat film (Eastman Kodak, Rochester, NY).
Statistical analysis
Data were expressed as mean ± standard error of the mean (SE). An analysis of variance (ANOVA) was performed with Statview 4.0 statistical analysis software (SAS Institute, Cary, NC), and a difference was accepted as significant if the P value was smaller than 0.05 as verified by the Bonferroni/ Dunn post hoc test.
RESULTS
Time course of liver HO-1 expression following LPS administration
Liver HO-1 expression in C57 mice after LPS administration was assessed by immunofluorescent staining and immunoblotting. HO-1 had low basal level expression in control liver by immunofluorescent staining (Figure 1A), but was barely detectable by immunoblotting (Figure 1B). A low dose (0.5 mg/kg) of LPS significantly increased HO-1 expression in the liver at 24 h after LPS administration. Furthermore, HO-1 expression was enhanced by three doses of LPS (Figure 1A and Figure 1B). The integrated intensity of HO-1 expression was significantly increased in liver of LPS-treated mice as compared with sham treatment mice (Figure 1C, P < 0.05). However, cellular HO-1 level went down if animals were allowed to recover for 48 h following the last LPS injection, then gradually declined to control level at 72 h (data not shown).
Figure 1.
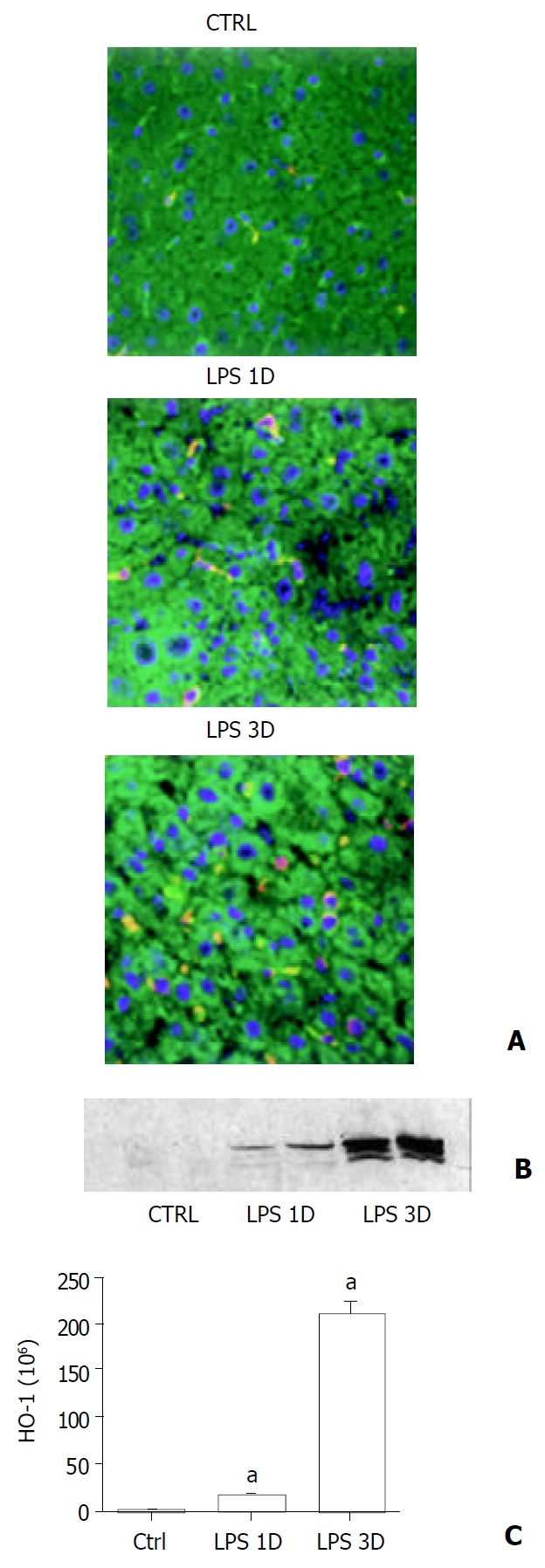
Liver HO-1 expression in wild type mice. A: Immun-ofluorescent detection of HO-1 in liver. After different doses of LPS or sham treatment, liver HO-1 expression of wild type (C57) mice were visualized by immunofluorescent staining with a specific rabbit polyclonal antibody against HO-1 fol-lowed by indocarbocyanine (Cy3)-conjugated anti-rabbit IgG (red). The cell surface was counterstained with fluorescein-con-jugated wheat germ agglutinin (green), and the nucleus was counterstained with bis-benzimide (blue). HO-1 was present in liver of sham-treated animals (Ctrl). A single dose of LPS treatment increased liver HO-1 expression (LPS 1D). Furthermore, HO-1 expression was enhanced by three doses of LPS treatment (LPS 3D, magnification ×400). B: Immunoblotting detection of HO-1 in liver. C57 mice were treated with vehicle (Ctrl), single dose of LPS (LPS 1D) and three doses of LPS (LPS 3D). Liver tissue was homogenized, and immunoblotting analysis was performed. HO-1 protein was detected by immunoblotting with polyclonal rabbit antibody against HO-1. Data were representative of at least 2 experiments. C: Im-munofluorescent staining to quantitate HO-1 expression. Liver HO-1 expression was determined by immunofluorescent staining. Integrated intensity of HO-1 positive signal was masked and quantified by using Slidebook software (I. I. I. Inc., Denver, CO). aP < 0.05 vs. sham.
Cell type involved in liver HO-1 expression
HO-1 protein was barely detectable in parenchymal liver cells following LPS stimulation, whereas high amounts of HO-1 were detectable in interstitial cells. To further characterize the cell types expressing HO-1, we attempted to localize the cell type(s) responsible for LPS-induced HO-1 expression in liver by double immunofluorescent staining. Rat anti-mouse CD68 monoclonal antibody and rabbit anti-HO-1 polyclonal antibody were used. We found that lower percentages of Kupffer cells exhibited positive staining for HO-1 in sham treatment. After three doses of LPS treatment, the majority of CD68 positive cells ( > 90%) were HO-1 positive (Figure 2).
Figure 2.
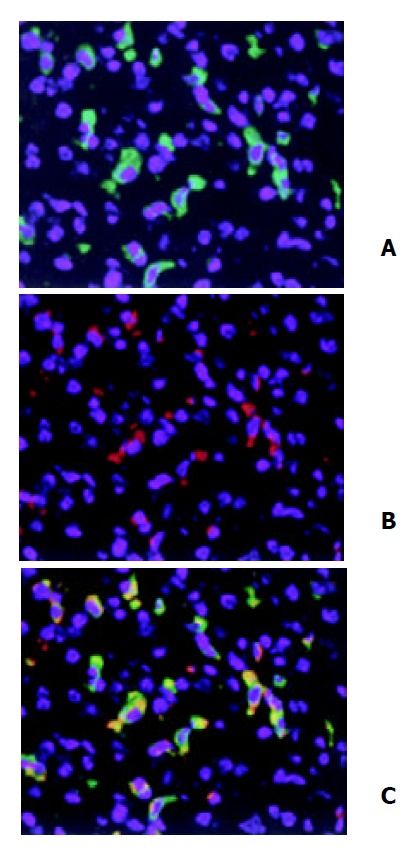
Double immunofluorescent staining of HO-1 local-ization in liver. After three doses of LPS treatment, HO-1 and CD68 in liver tissue were detected by double immunofluores-cent staining with polyclonal rabbit antibody against HO-1 followed by FITC-conjugated anti-rabbit IgG (green) and mono-clonal rat antibody against mouse CD68 followed by Cy3-con-jugated anti-rat IgG (red). Cell nuclei were counterstained with bis-benzimide (blue). A: HO-1; B: Macrophages (Kupffer cells); C: HO-1 + Macrophages. magnification × 400.
Effect of TLR4 on liver HO-1 expression
To examine the mechanism of regulation of liver HO-1 expression in response to LPS, we used C.C3H-Tlr4Lps-d mice, mice of TLR4 mutation. In BALB/cJ (wild type) mice, LPS-induced HO-1 expression in liver was comparable to C57 mice. In contrast, liver HO-1 expression in TLR4 mutant mice was significantly inhibited following the same treatment with LPS (Figure 3). These results suggested that mutation of TLR4 exerted a dominant effect on liver HO-1 expression after LPS treatment.
Figure 3.
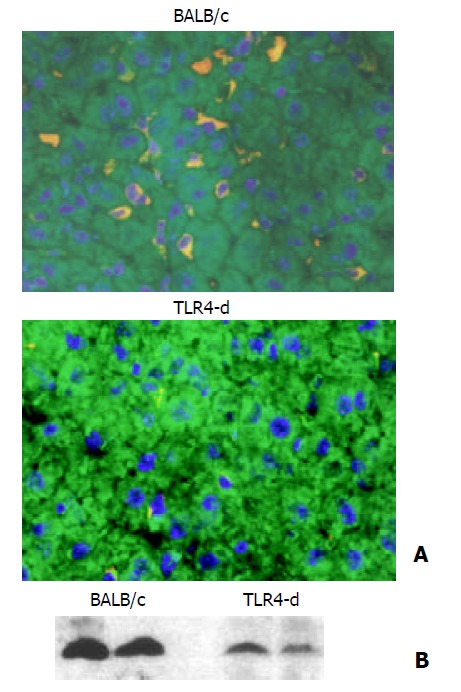
Effect of TLR4 mutation on liver HO-1 expression. A: Immunofluorescent detection of HO-1 in liver. After three doses of LPS treatment, liver HO-1 expression was visualized by immunofluorescent staining with a specific rabbit polyclonal antibody against HO-1, followed by indocarbocyanine (Cy3)-conjugated anti-rabbit IgG (red). The cell surface was counter-stained with fluorescein-conjugated wheat germ agglutinin (green), and the nucleus was counterstained with bis-benzimide (blue). Liver HO-1 expression was abrogated in TLR4 mutated mice following LPS, but neither was influenced in BALB/c (control) mice (magnification ×400). B: Immunoblotting detection of HO-1 in liver. BALB/c and TLR4 mutated mice were treated with three doses of LPS. Liver tissue was homogenized, and immunoblotting analysis was performed. HO-1 protein was detected by immunoblotting with polyclonal rabbit antibody against HO-1. Data were repre-sentative of at least 2 experiments.
Effect of TNF knockout on HO-1 expression
LPS is a potent inducer of TNF-α. Recent evidence suggests that LPS-induced TNF-α production is mediated through TLR4. Previous study demonstrated that TNF-α induced HO-1 expression in endothelial cells. Does TNF-α play an important role in LPS-induced HO-1 expression in liver To examine the mechanism of regulation of liver HO-1 expression in response to LPS, we used C.C3H-Tlr4Lps-d mice, mice of TLR4 mutation. In BALB/cJ (wild type) mice, LPS-induced HO-1 expression in liver was comparable to C57 mice. In contrast, liver HO-1 expression in TLR4 mutant mice was significantly inhibited following same treatment as wild type mice (Figure 3). These results suggested that TLR4 exerted a dominant effect on liver HO-1 expression following LPS treatment.
Effect of IL-1β knockout on HO-1 expression
IL-1β is characteristically present in many inflammatory disorders. Although previous studies showed that IL-1β induced HO-1 expression in vitro, little is known about the role of endogenous IL-1β in LPS-induced HO-1 expression. We determined liver HO-1 expression in IL-1β KO mice. Mice were treated with LPS same as C57 mice. Compared with C57 mice, mice of IL-1β KO showed only a slight difference in liver HO-1 expression following LPS treatment (Figure 4), indicating that the expression of HO-1 following LPS was partially mediated by endogenous IL-1β.
Figure 4.
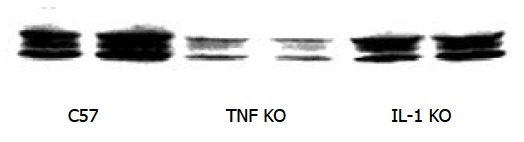
Effect of TNF KO and IL-1 KO on liver HO-1 expression. Immunoblotting detection of HO-1 in liver. C57, TNF KO and IL-1 KO mice were treated with three doses of LPS. Liver tissue was homogenized, and immunoblotting analysis was performed. HO-1 protein was detected by immunoblotting with polyclonal rabbit antibody against HO-1. Data were representative of at least 2 experiments.
DISCUSSION
In this study, we found that a low dose of LPS significantly increased HO-1 expression only in macrophages of wild type mice. This effect was further enhanced by the same dose of LPS for 3 consecutive days. However, HO-1 expression was significantly inhibited in TLR4 mutant mice. While liver HO-1 expression in TNF KO mice was much lower than that in C57 mice following the same LPS treatment, IL-1 KO had a slight influence on liver HO-1 expression following LPS treatment. The present results confirmed that macrophages were the major source of HO-1 in the lungs and liver as induced by LPS. This study demonstrated that TLR4 played a dominant role in mediating HO-1 expression following LPS. LPS-induced HO-1 expression was mainly mediated by endogenous TNF, but only partially by endogenous IL-1.
HO-1 is the rate-limiting enzyme in heme catabolism, and is also known as heat shock protein 32. Previous studies demonstrated that HO-1, besides its role in heme degradation, may also play a vital function in maintaining cellular homeostasis[26]. HO-1 is induced not only by its substrate, hemin, but also by oxidative stress, heat stress, UV irradiation, heavy metals, LPS and cytokines[4-8]. It has also been reported that HO-1 is induced in liver after LPS administration[11,13,14]. But most of studies just determined the changes of HO-1 mRNA in liver. Little is known about the time course and localization of HO-1 protein expression in liver. We examined liver HO-1 expression following LPS. We found that HO-1 had a low basal level expression in normal mouse liver by immunofluorescent staining. A low dose of LPS significantly increased HO-1 expression in the liver at 24 h after LPS administration. HO-1 expression was enhanced by three doses of LPS. Induction of HO-1, however, went down when animals were allowed to recover 48 h following the last LPS injection, then gradually declined to the control level by 72 h (data not shown). These results indicate that LPS-induced HO-1 expression in liver is in a dose-dependent fashion. Furthermore, we detected and localized HO-1 using immunofluorescent double staining. Hepatocytes were involved in the removal of plasma hemoglobin and some studies suggested that LPS mediated HO-1 mRNA accumulation in hepatocytes[7,13]. Interestingly, in the present study, most of HO-1 positive signals were co-localized in Kupffer cells. This finding formed a contrast to those from other animal models by hemin and LPS treatment. It is possible that the differences of experimental design, i.e, different stimuli, doses, and animals may lead to different results. In addition, the method of HO-1 staining by others was different from ours, it could be a reason for different expression of HO-1 in liver. Kupffer cells are the first line defense and effector cells in liver inflammatory response. The present results confirm that Kupffer cells are the major source of HO-1 in liver induced by LPS. HO-1 expression in Kupffer cells may play a protective role by inducing an adaptive hepatocellular stress response after LPS stimulation.
Studies demonstrated that LPS induced a cellular response by the signaling molecules belonging to the Toll-like receptor family[15]. TLR4 has been shown to be essential for the recognition of LPS, lipoteichoic acid (LTA) and Taxol[17-19]. Recently, it was demonstrated that TLR4 mediated heat shock protein 60 signaling[20]. Although TLR4 is clearly critical to LPS signaling, the role of TLR4 in LPS-induced HO-1 expression has remained unclear. To examine the mechanism of regulation of liver HO-1 expression in response to LPS, we determined liver HO-1 expression in TLR4 mutant mice. Liver HO-1 expression in TLR4 mutant mice was significantly inhibited following the same treatment as wild type mice. This result suggests that TLR4 exerts a dominant effect on liver HO-1 expression after LPS treatment.
TNF-α is a pleiotropic early response cytokine and is rapidly produced by LPS stimulation. To determine the role of TNF-α in HO-1 expression following LPS in vivo, we subjected TNF KO mice to the same treatment as C57 mice. This treatment could also result in liver HO-1 expression comparable to that in the liver of C57 mice. We found that liver HO-1 expression in TNF KO mice was much lower than that in C57 mice following the same LPS treatment. This result suggests that LPS induces HO-1 expression mainly through endogenous TNF.
As an important inflammatory cytokine, IL-1 (IL-1α and IL-1β) induces HO-1 expression in endothelial cells and pancreatic islets[23-28]. Toll-like receptors belong to the IL-1 receptor family containing repeated leucine-rich motifs in their extracellular portion and are linked to a signaling pathway that involves the IL-1-receptor-associated kinase and NF-κB. It is likely that IL-1-induced HO-1 expression is mediated by TLR4. But it is unclear about the role of endogenous IL-1 in LPS-induced liver HO-1 expression. So, in the present study, IL-1 KO mice were used to determine liver HO-1 expression following LPS induction. Mice were treated with LPS the same as C57 mice. Compared with C57 mice, IL-1β KO has a slight influence on liver HO-1 expression following LPS treatment of IL-1β KO mice, indicating that the expression of HO-1 following LPS is partially mediated by endogenous IL-1β.
Taken together, the current study in vivo showed that TLR4 mutation attenuated LPS-induced HO-1 production in tissues. This provides a strong support to the hypothesis that TLR4 plays an important role in HO-1 signals. However, some aspects of its action remain unknown. What are the down stream signals of TLR4 In this context, future investigations are necessary to determine the signal transduction pathway of HO-1 expression induced by LPS.
Footnotes
Edited by Lu HM and Wang XL
References
- 1.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 2.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- 3.Otterbein LE, Choi AM. Heme oxygenase: colors of defense against cellular stress. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1029–L1037. doi: 10.1152/ajplung.2000.279.6.L1029. [DOI] [PubMed] [Google Scholar]
- 4.Camhi SL, Alam J, Otterbein L, Sylvester SL, Choi AM. Induction of heme oxygenase-1 gene expression by lipopolysaccharide is mediated by AP-1 activation. Am J Respir Cell Mol Biol. 1995;13:387–398. doi: 10.1165/ajrcmb.13.4.7546768. [DOI] [PubMed] [Google Scholar]
- 5.Choi AM, Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol. 1996;15:9–19. doi: 10.1165/ajrcmb.15.1.8679227. [DOI] [PubMed] [Google Scholar]
- 6.Keyse SM, Tyrrell RM. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci USA. 1989;86:99–103. doi: 10.1073/pnas.86.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otterbein L, Sylvester SL, Choi AM. Hemoglobin provides protection against lethal endotoxemia in rats: the role of heme oxygenase-1. Am J Respir Cell Mol Biol. 1995;13:595–601. doi: 10.1165/ajrcmb.13.5.7576696. [DOI] [PubMed] [Google Scholar]
- 8.Terry CM, Clikeman JA, Hoidal JR, Callahan KS. Effect of tumor necrosis factor-alpha and interleukin-1 alpha on heme oxygenase-1 expression in human endothelial cells. Am J Physiol. 1998;274:H883–H891. doi: 10.1152/ajpheart.1998.274.3.H883. [DOI] [PubMed] [Google Scholar]
- 9.Camhi SL, Alam J, Wiegand GW, Chin BY, Choi AM. Transcriptional activation of the HO-1 gene by lipopolysaccharide is mediated by 5' distal enhancers: role of reactive oxygen intermediates and AP-1. Am J Respir Cell Mol Biol. 1998;18:226–234. doi: 10.1165/ajrcmb.18.2.2910. [DOI] [PubMed] [Google Scholar]
- 10.Carraway MS, Ghio AJ, Taylor JL, Piantadosi CA. Induction of ferritin and heme oxygenase-1 by endotoxin in the lung. Am J Physiol. 1998;275:L583–L592. doi: 10.1152/ajplung.1998.275.3.L583. [DOI] [PubMed] [Google Scholar]
- 11.Oshiro S, Takeuchi H, Matsumoto M, Kurata S. Transcriptional activation of heme oxygenase-1 gene in mouse spleen, liver and kidney cells after treatment with lipopolysaccharide or hemoglobin. Cell Biol Int. 1999;23:465–474. doi: 10.1006/cbir.1999.0375. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki T, Takahashi T, Yamasaki A, Fujiwara T, Hirakawa M, Akagi R. Tissue-specific gene expression of heme oxygenase-1 (HO-1) and non-specific delta-aminolevulinate synthase (ALAS-N) in a rat model of septic multiple organ dysfunction syndrome. Biochem Pharmacol. 2000;60:275–283. doi: 10.1016/s0006-2952(00)00324-5. [DOI] [PubMed] [Google Scholar]
- 13.Bauer I, Wanner GA, Rensing H, Alte C, Miescher EA, Wolf B, Pannen BH, Clemens MG, Bauer M. Expression pattern of heme oxygenase isoenzymes 1 and 2 in normal and stress-exposed rat liver. Hepatology. 1998;27:829–838. doi: 10.1002/hep.510270327. [DOI] [PubMed] [Google Scholar]
- 14.Rizzardini M, Zappone M, Villa P, Gnocchi P, Sironi M, Diomede L, Meazza C, Monshouwer M, Cantoni L. Kupffer cell depletion partially prevents hepatic heme oxygenase 1 messenger RNA accumulation in systemic inflammation in mice: role of interleukin 1beta. Hepatology. 1998;27:703–710. doi: 10.1002/hep.510270311. [DOI] [PubMed] [Google Scholar]
- 15.Means TK, Golenbock DT, Fenton MJ. The biology of Toll-like receptors. Cytokine Growth Factor Rev. 2000;11:219–232. doi: 10.1016/s1359-6101(00)00006-x. [DOI] [PubMed] [Google Scholar]
- 16.Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J Immunol. 2000;164:5564–5574. doi: 10.4049/jimmunol.164.11.5564. [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki K, Akashi S, Shimazu R, Yoshida T, Miyake K, Nishijima M. Mouse toll-like receptor 4.MD-2 complex mediates lipopolysaccharide-mimetic signal transduction by Taxol. J Biol Chem. 2000;275:2251–2254. doi: 10.1074/jbc.275.4.2251. [DOI] [PubMed] [Google Scholar]
- 18.Means TK, Lien E, Yoshimura A, Wang S, Golenbock DT, Fenton MJ. The CD14 ligands lipoarabinomannan and lipopolysaccharide differ in their requirement for Toll-like receptors. J Immunol. 1999;163:6748–6755. [PubMed] [Google Scholar]
- 19.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 20.Ohashi K, Burkart V, Flohé S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 21.Means TK, Jones BW, Schromm AB, Shurtleff BA, Smith JA, Keane J, Golenbock DT, Vogel SN, Fenton MJ. Differential effects of a Toll-like receptor antagonist on Mycobacterium tuberculosis-induced macrophage responses. J Immunol. 2001;166:4074–4082. doi: 10.4049/jimmunol.166.6.4074. [DOI] [PubMed] [Google Scholar]
- 22.Nill MR, Oberyszyn TM, Ross MS, Oberyszyn AS, Robertson FM. Temporal sequence of pulmonary cytokine gene expression in response to endotoxin in C3H/HeN endotoxin-sensitive and C3H/HeJ endotoxin-resistant mice. J Leukoc Biol. 1995;58:563–574. doi: 10.1002/jlb.58.5.563. [DOI] [PubMed] [Google Scholar]
- 23.Terry CM, Clikeman JA, Hoidal JR, Callahan KS. TNF-alpha and IL-1alpha induce heme oxygenase-1 via protein kinase C, Ca2+, and phospholipase A2 in endothelial cells. Am J Physiol. 1999;276:H1493–H1501. doi: 10.1152/ajpheart.1999.276.5.H1493. [DOI] [PubMed] [Google Scholar]
- 24.Ye J, Laychock SG. A protective role for heme oxygenase expression in pancreatic islets exposed to interleukin-1beta. Endocrinology. 1998;139:4155–4163. doi: 10.1210/endo.139.10.6244. [DOI] [PubMed] [Google Scholar]
- 25.Song Y, Ao L, Calkins CM, Raeburn CD, Harken AH, Meng X. Differential cardiopulmonary recruitment of neutrophils during hemorrhagic shock: a role for ICAM-1. Shock. 2001;16:444–448. doi: 10.1097/00024382-200116060-00007. [DOI] [PubMed] [Google Scholar]
- 26.Horváth I, MacNee W, Kelly FJ, Dekhuijzen PN, Phillips M, Döring G, Choi AM, Yamaya M, Bach FH, Willis D, et al. "Haemoxygenase-1 induction and exhaled markers of oxidative stress in lung diseases", summary of the ERS Research Seminar in Budapest, Hungary, September, 1999. Eur Respir J. 2001;18:420–430. doi: 10.1183/09031936.01.00231201. [DOI] [PubMed] [Google Scholar]
- 27.Kyokane T, Norimizu S, Taniai H, Yamaguchi T, Takeoka S, Tsuchida E, Naito M, Nimura Y, Ishimura Y, Suematsu M. Carbon monoxide from heme catabolism protects against hepatobiliary dysfunction in endotoxin-treated rat liver. Gastroenterology. 2001;120:1227–1240. doi: 10.1053/gast.2001.23249. [DOI] [PubMed] [Google Scholar]
- 28.West MA, Bennet T, Seatter SC, Clair L, Bellingham J. LPS pretreatment reprograms macrophage LPS-stimulated TNF and IL-1 release without protein tyrosine kinase activation. J Leukoc Biol. 1997;61:88–95. doi: 10.1002/jlb.61.1.88. [DOI] [PubMed] [Google Scholar]