

Significance
MicroRNAs can repress target genes to regulate cellular function in vivo. The miR-17∼92 cluster is critical for cell cycle control, survival signaling, and angiogenesis, but the individual components of the cluster and their endogenous roles in vivo are less well understood. Here we show that the loss of endothelial-derived miR-17∼92 increases basal cardiac and limb arteriogenesis and improves ischemia-induced changes in blood flow. One component of the cluster, miR-19, directly targets FZD4 and LRP6, thereby regulating WNT signaling in vitro and in vivo, and antagonism of miR-19 up-regulates WNT signaling and improves defective blood flow recovery.
Keywords: endothelium, arteriogenesis, vascular, microRNA
Abstract
The contribution of endothelial-derived miR-17∼92 to ischemia-induced arteriogenesis has not been investigated in an in vivo model. In the present study, we demonstrate a critical role for the endothelial-derived miR-17∼92 cluster in shaping physiological and ischemia-triggered arteriogenesis. Endothelial-specific deletion of miR-17∼92 results in an increase in collateral density limbs and hearts and in ischemic limbs compared with control mice, and consequently improves blood flow recovery. Individual cluster components positively or negatively regulate endothelial cell (EC) functions in vitro, and, remarkably, ECs lacking the cluster spontaneously form cords in a manner rescued by miR-17a, -18a, and -19a. Using both in vitro and in vivo analyses, we identified FZD4 and LRP6 as targets of miR-19a/b. Both of these targets were up-regulated in 17∼92 KO ECs compared with control ECs, and both were shown to be targeted by miR-19 using luciferase assays. We demonstrate that miR-19a negatively regulates FZD4, its coreceptor LRP6, and WNT signaling, and that antagonism of miR-19a/b in aged mice improves blood flow recovery after ischemia and reduces repression of these targets. Collectively, these data provide insights into miRNA regulation of arterialization and highlight the importance of vascular WNT signaling in maintaining arterial blood flow.
MicroRNAs (miRNAs) are regulatory small noncoding RNAs with a key role in regulating multiple pathways involved in development and disease. These 20–25 nucleotides function via posttranscriptional repression of targeted gene expression containing complementary sites in the 3′ untranslated region (UTR), resulting in degradation of the targeted mRNA or inhibition of its translation (1). Given the myriad of potential targets of miRNAs, identifying the dominant functions of specific miRNAs in the context of development, physiology, and disease is challenging and important for the realization of miRNA therapeutics.
The miRNA17∼92 cluster, a polycistronic miRNA that produces six individual miRNAs (miR-17, miR-18a, miR-19a, miR-19b, miR-20a, and miR-92a), has been demonstrated to exhibit various biological functions regulating multiple cellular processes involved in proliferation, oncogenesis, differentiation, angiogenesis, and survival (2–5). The region encoding for this cluster, C13orf25, is amplified in multiple types of lymphomas, including diffuse large B-cell lymphoma, follicular lymphoma, and some solid tumors (6). Its wide spectrum of validated targets implies effects on multiple signaling pathways, such as BMP, TGF, and PI3K signaling involved in development and disease (7–12). It is well known that individual miRNAs of this cluster can cooperate or work independently to efficiently modulate multiple signaling pathways, as has been demonstrated for miR-19, which drives the oncogenic ability of this cluster in the context of a MYC-driven B-cell lymphoma (9).
Although comprehensive lists of potential targets of the miR-17∼92 cluster have been generated, examination of the role of this cluster and validation of targets in the context of vascular biology have been limited. Initial work demonstrated that vascular endothelial growth factor (VEGF) and shear stress can induce components of the cluster in endothelial cells (ECs) (3, 13). The contribution of EC-derived miR-17∼92 to angiogenesis and ischemia-induced arteriogenesis has not yet been investigated in an in vivo model. Emerging evidence from studies using systemic injections of antagomir suggests that miR-92a controls functional recovery of damaged tissue in mouse models of myocardial infarction and hindlimb ischemia (HLI), implying that miR-92a targets multiple proangiogenic proteins, including integrin alpha-5 (14), and coregulates KLF2 and KLF4, suggesting a role in atherogenesis (15). Other components of the cluster, such as miR-17, have been demonstrated to play an antiangiogenic role in ECs (16), whereas in the context of Myc-induced Ras-transformed colonocyte cells, miR-18a and miR-19a have been shown to target antiangiogenic proteins CTGF and Tsp1 (4).
The WNT signaling pathway plays multiple roles in governing fundamental mechanisms of cell proliferation, cell polarity, and cell fate determination during embryonic development and tissue homeostasis (17). The canonical WNT pathway transduces its activity via activation of the seven transmembrane Frizzled receptors (FZD) in complex with its coreceptor of the low-density lipoprotein receptor-related protein 5/6 class (LRP5/6). The formation of a WNT-FZD-LRP6 complex, together with the recruitment of scaffolding proteins, results in stabilization of cytosolic β-catenin, which accumulates and translocates to the nucleus to form complexes with TCF/LEF and activate WNT target gene expression (17). Aberrant activation of components of this pathway in ECs may produce severe vascular phenotypes and result in embryonic lethality in some cases (18–23). Of relevance, FZD4 and LRP5, which has high homology to LRP6 and shares many biochemical properties (24), have been linked to vascular disorders, including defective intraretinal and cerebellar vascular patterning and arterial network formation (21, 23, 25).
In the present study, we used endothelial-specific miR-17∼92 loss-of-function mutant mice to directly assess the physiological and cellular contributions of this cluster to EC function during pathological states of arteriogenesis. Indeed, 17∼92 EC KO mice exhibit accelerated blood flow recovery and enhanced arterial vessel density after limb ischemia. Interestingly, 17∼92 EC KO mice have greater numbers of hindlimb and coronary arterial vessels in the absence of ischemia, implying that the endogenous cluster is critical for developmental arteriogenesis/collateral vessel genesis. Mechanistically, the loss of miR-17∼92 induces enhanced EC morphogenesis, an effect rescued by miR-17, miR-18a, and miR-19a, and we show that miR-19a directly targets both FZD4 and LRP6. Treatment of aged mice with defective ischemic recovery with locked nucleic acid (LNA) miR-19 phenocopies the accelerated blood flow recovery observed in the miR-17∼92 EC-specific KO mice, and increases tissue levels of FZD4 and LRP6. Collectively, these data support the concept that the miR-17∼92 cluster, and in particular miR-19, physiologically represses arteriogenesis, in part by suppressing the WNT signaling pathway by targeting FZD4 and LRP6.
Results
Characterization of Endothelial-Specific miR-17∼92 KO Mice.
To elucidate the role of endothelial-derived miR17∼92 in EC physiology, we crossed mice harboring a floxed miR-17∼92 allele with transgenic mice expressing Cre under the control of Tie2 or a tamoxifen-inducible CDH5 (VE-cadherin) promoter (5, 26, 27). To assess the level of inactivation of the miR-17∼92 cluster, we isolated ECs from the lungs of 17∼92fl/fl (WT) and Tie2-Cre;17∼92fl/fl animals (17∼92 EC KOTie2), cultured and processed for quantitative RT-PCR (qRT-PCR) of cluster components (Fig. S1). Both the primary (pri)-miR-17∼92 transcript and mature components of the cluster were reduced by >90% (Fig. S1 A and B), whereas expression of the miR17∼92 paralog, pri-miR-106b∼25 cluster, and the EC-enriched miRNA, miR-126-3p, was not affected (Fig. S1 A and B, respectively). The additional X-linked paralog cluster, pri-miR-106a-363, was not detectable in ECs. Similar findings were observed in primary ECs isolated from hearts of tamoxifen-inducible VE cadherin Cre mice (VE-Cad-Cre-ERT2;17∼92fl/fl), referred to as 17∼92 EC KOVE-Cad (Fig. S1C). The loss of miR-17∼92 in ECs was not embryonic- lethal; however, EC-specific deletion did reduce the body weight of the mice using both Cre driver lines (Fig. S2).
Fig. S1.
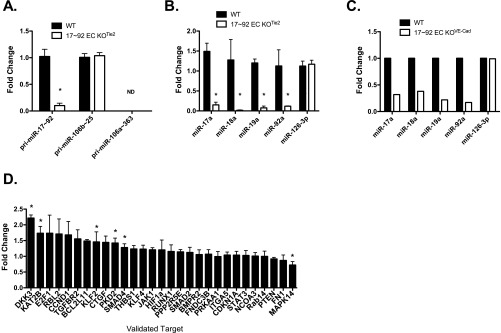
Characterization of miR-17∼92 expression and its validated target gene expression in primary ECs derived from 17∼92 EC KOTie2 and 17∼92 EC KOVE-Cad mice. (A and B) qRT-PCR analysis of cultured primary ECs demonstrating a marked reduction of precursor miR-17∼92 (pri) and its mature derivatives. Data are normalized to WT primary ECs and represent the average of three independent isolations. *P < 0.05. (C) qRT-PCR of primary ECs derived from 17∼92 EC KOVE-Cad animals for detection of mature components of miR-17∼92 and its paralogs. Data are normalized to WT primary ECs and are derived from freshly isolated heart ECs pooled from three animals per group. (D) Expression of validated targets of miR-17∼92 examined via qRT-PCR in primary ECs from WT and 17∼92 EC KOTie2 mice. Data represent fold change compared with primary WT ECs and are derived from at least three independent isolations. *P < 0.05.
Fig. S2.

Body weight is reduced in 17∼92 EC KOTie2 and 17∼92 EC KOVE-Cad mice compared with WT mice. Deletion in 17∼92 EC KOVE-Cad was induced at postnatal day 1. n = 4–11 animals per group. *P < 0.05, two-way ANOVA.
To examine the functional importance of the endogenous cluster in repressing gene expression, we measured mRNA levels of known, validated target genes in primary lung ECs isolated and cultured in the presence of serum and EC growth factors from WT and 17∼92 EC KOTie2 mice. Validated genes were defined as genes with mRNA levels reduced by cluster mimic miRNAs, up-regulated by anti-miRs. and shown to be direct targets via 3′ UTR–luciferase constructs. The loss of miR-17∼92 in ECs increased the levels of some (Dkk3, KAT2b, BCL2L11, PKD2, SMAD4, and JAK1), but not all, “validated” targets (Fig. S1D), implying that the cluster may exert cell specific repression of targets, or that the activation state of the cell is critical for miRNA regulation of its putative target genes.
Loss of miR-17∼92 Stimulates Arteriogenesis After Limb Ischemia.
The systemic administration of an antagomir designed to inhibit miR-92a improves perfusion and functional recovery in an HLI model by targeting the expression of multiple angiogenic proteins, including the integrin alpha-5 subunit (14). Given that the miR-17∼92 cluster encodes for six mature miRNAs that are coordinately expressed and may act independently, cluster components likely regulate mRNA levels or translation that may modulate arteriogenesis. To examine a role for the endothelial-derived cluster in HLI, we subjected aged (6 mo old) male mice to HLI and measured blood flow using a deep-penetrating laser Doppler probe as described previously (28). EC-specific deletion of the miR-17∼92 cluster improved functional recovery from HLI (Fig. 1A). We obtained similar results, albeit to a lesser extent, using the inducible VE-Cad Cre model and postnatal deletion (Fig. S3). This postnatal strategy typically results in reduced deletion efficiency.
Fig. 1.

Loss of miR-17∼92 in ECs improves blood flow recovery after HLI. (A) Laser Doppler analysis was used to measure blood flow in the gastrocnemius muscle before, immediately after, and at 3, 7, and 14 d after HLI. n = 9–10 per group. *P < 0.05, two way ANOVA. (B) Reconstructed micro-CT images of arterial vessels of WT and 17∼92 EC KOTie2 mice at 14 d post-HLI. Some 30% of the 17∼92 EC KOTie2 mice had an alternative major collateral artery that was obvious immediately on arteriectomy of the femoral artery, as depicted in C (arrow). (C and D) Quantitative analysis of micro-CT images in the upper and lower limb under ischemic (C) and nonischemic contralateral limbs (D). Data are presented as total number of vascular structures in z-axis slices. n = 4 mice per group. *P < 0.05, two-way ANOVA. All data are mean ± SEM.
Fig. S3.

miR-17∼92 deletion in 17∼92 EC KOVE-Cad mice improves HLI blood flow recovery. Deletion was induced at 2 wk before surgery. n = 5–7 per group. *P < 0.05, two-way ANOVA.
Based on the rapid blood flow recovery in the EC-specific KO lines, we speculated that collateral formation via arteriogenesis may be augmented in mice lacking the 17∼92 cluster in ECs. We quantified the extent of arteriogenesis using 3D micro-computed tomography (micro-CT) imaging. Mice were perfused with vasodilators and contrast agents to assess arterial density, but not the density of capillaries or veins. Loss of the 17∼92 cluster in ECs revealed a robust increase in collateral vasculature density (<48 μm) in ischemic limbs compared with control mice (Fig. 1 B and C). Interestingly, 30% of these mice had an alternative major collateral artery that was functional and perfused immediately on arteriectomy of the femoral artery (Fig. 1B, Far Right; arrow denotes unique vessel). This adaptive response was not observed in any of the control mice examined, or in any of the hundreds of mice that we have examined in the past, indicative of a 30% penetrant arteriogenic phenotype for the cluster-deficient mice.
To quantify baseline arteriogenesis in the absence of ischemia, we performed micro-CT imaging of the arterial vasculature of the contralateral, nonischemic limb. Surprisingly, mice lacking the cluster in ECs revealed an arterial phenotype with a significantly greater arteriole density in thigh vessels with a diameter of ≤72 μm (Fig. 1D). This finding suggests that the EC miR-17∼92 cluster is important for the proper development of hindlimb arteries and collaterals.
To further elucidate the contribution of this cluster to arterial development independent of ischemia, we performed micro-CT imaging of the arterial vasculature in limbs and hearts of aged (6 mo old) WT and 17∼92 EC KOTie2 mice. The arterial density of vessels with a diameter of ≤48 μm in the thigh (Fig. 2A) and ≤16 μm in the heart (Fig. 2 B and C) was more abundant in 17∼92 EC KOTie2 mice compared with littermate control mice. These data suggest that the 17∼92 cluster represses physiological processes that govern the extent of baseline and ischemic arteriogenesis in the heart and limbs.
Fig. 2.

Mice lacking endothelial miR-17∼92 display enhanced arterial density of small arteries in limbs (A: thigh, Left; calf, Right) and heart (B and C). (B) Reconstructed micro-CT images of hearts of WT (Left) and 17∼92 EC KOTie2 (Right) mice show increased arteriole density in heart vessels with a diameter of ≤16 μm. Data are mean ± SEM; n = 4 mice per group. *P < 0.05, two-way ANOVA.
Loss of miR-17∼92 in ECs Influences Functions in Vitro.
We next analyzed several aspects of EC function—growth, adhesion, and cord formation—in ECs isolated from WT and 17∼92 EC KOTie2 mice. ECs lacking the 17∼92 cluster grew more slowly, as measured by BrdU incorporation, consistent with the known function of the cluster in promoting cell growth via repression of E2F or PTEN (Fig. 3A) (2, 4, 9, 29–31). ECs lacking the miR-17∼92 cluster demonstrated augmented adhesion to either gelatin or fibronectin, complementing data showing that overexpression of the cluster reduces adhesion (Fig. 3B) (8) and that 17∼92 KO ECs exhibit enhanced cord formation both under basal conditions and when stimulated with bFGF (Fig. 3C) (14, 16). In addition, EC migration into wounded monolayers (Fig. S4 A and B) and aortic sprouting (Fig. S4 C and D) are augmented in miR-17∼92 KO ECs and vessels, respectively. These data support the idea that the net effect of loss of the miR-17∼92 cluster in ECs reduces growth but promotes adhesion and sprouting.
Fig. 3.

Characterization of EC phenotypes in ECs from WT and 17∼92 KO mice. (A) BrdU incorporation was used to evaluate proliferation of WT and 17∼92 KO ECs under 0.5% FBS or 20% FBS. *P < 0.05. (B) Adhesion of ECs from both groups was assessed at 30 min after seeding on uncoated, gelatin-coated, or fibronectin-coated plates. *P < 0.05. (C) Cord formation of WT and 17∼92 KO ECs was examined under basal conditions (0.5% FBS) or with bFGF (10 ng/mL) as a stimulant. The 17∼92 KO cells exhibited augmented cord formation under both basal and stimulated conditions. Data are mean ± SEM and are representative of an experiment repeated three additional times and conducted in triplicate. (Scale bar: 200 μm.)
Fig. S4.

(A and B) Loss of miR 17∼92 KO in ECs augments recovery after wounding (A) and aortic sprouting (B) in vitro. Primary WT or 17∼92 KO ECs were starved, then wounded and treated with DMEM plus 0.5% FBS or 10 ng/mL bFGF2 for 12 h. The wound area was measured in captured images at 0 and 12 h, and migration was quantified. Data are duplicates from two experiments. (C) Aortic rings from WT and 17∼92VE-Cad mice were embedded in collagen type 1 and treated with +2.5% (vol/vol) FBS or VEGF 100 ng/mL for 6 d. At day 6, rings were fixed with PFA (4%) and stained with BS1 Lectin-FITC to delineate ECs. (D) Images of ring sprouting were obtained and quantified. Data are representative of five or six mice per group, and sprouts from eight or nine rings per mouse were quantified. Error bars represent mean ± SEM. *P < 0.05, one-way ANOVA with Bonferroni’s posttest. Data are representative of an experiment repeated three times and conducted in triplicate. *P < 0.05.
WNT-Related Receptor FZD4 and LRP6 Are Targeted by the miR-17∼92 Cluster.
The enhanced baseline and stimulated arteriogenesis in mice lacking the miR-17∼92 cluster in ECs is reminiscent of a recent report of reduced basal and ischemia-stimulated arterial density in mice lacking the WNT receptor FZD4 (25). In addition, ECs derived from these FZD4 KO mice failed to form cords in vitro (21, 25). Given the inverse correlations between the phenotypes that we observed and the vascular phenotypes in the FZD4 KO mice, we examined whether endothelial-derived miR-17∼92 can potentially regulate FZD4 and the signaling pathway downstream of its activation.
Based on in silico analysis, the WNT-related receptor FZD4 and it coreceptor LRP6 have putative target sites in the 3′ UTR for components of the cluster (Fig. S5). Thus, we compared FZD4 and LRP6 mRNA levels in WT and 17∼92 KO ECs. qRT-PCR expression analysis showed significantly higher mRNA levels of both FZD4 and LRP6 in 17∼92 KO ECs compared with WT ECs (Fig. 4A). In our experiments, antibodies for the detection of endogenous mouse FZD4 protein did not work in Western blot analyses using siRNA to FZD4 as a positive control for specificity; however, we did confirm the higher LRP6 protein levels in 17∼92 KO ECs (Fig. 4B).
Fig. S5.

LRP6 and FZD4 are functionally relevant miR-19 target genes. (A) Schematic representation of the sequences of the miR-19 predicted binding sites in the 3′ UTR of FZD4 and LRP6. Mutation sites are marked with an asterisk. Sequence differences between miR19a and miR19b are indicated by “_”. (B) Mutation of predicted miR-19a target sites reduces miR-19a– mediated repression of 3′ UTR LUC activity of FZD4 and LRP6. Data are mean ± SEM from three independent experiments.
Fig. 4.

FZD4 and LRP6 are direct miR-19 targets. (A) qRT-PCR analysis showing enhanced mRNA expression of FZD4 and LRP6 in 17∼92 KO ECs compared with WT ECs. Data are from four independent isolations. (B) Expression levels of LRP6 protein in ECs isolated from 17∼92 KO and WT mice. (C) Changes in mRNA expression levels of FZD4 and Lrp6 in 17∼92 KO ECs following transfection with the indicated mimics (30 nM). (D) Cotransfection of miR-19a (60 nM) significantly reduced 3′ UTR luciferase reporter activity for FZD4 and LRP6 measured in HEK293 cells. The data shown are representative of an experiment repeated three times and conducted in triplicate. (E and F) Primary ECs were treated with either control CM (labeled –) or WNT3a CM for 5 h or for 2 or 5 h. Fractionated cytoplasmic (E) and nuclear (F) samples were analyzed for protein stabilization of β-catenin. Data are quantified below the blots relative to loading controls and are representative of an experiment repeated three times. (G) BAT-gal reporter for WNT/β-catenin signaling was examined in WT/BAT-gal or 17∼92 EC KOVE-Cad/BAT animals. Adult mice were killed at 2 wk after tamoxifen induction of cluster deletion, and mesenteric arteries were triple-stained en face for β-gal (green) to detect β-catenin–induced expression, for PECAM (red) and DAPI. Double-staining for β-gal and PECAM was detected in the nuclei of 17∼92 KO animals; n = 3–4. (Scale bar: 25 μm.) (H) mRNA expression of WNT3a transcriptionally regulated genes as analyzed by qRT-PCR. ECs were transfected with scramble or mimic miR-19a (30 nM). At 48 h after transfection, cells were serum-starved for 3 h and then stimulated with control CM or WNT3a CM for 2–6 h. Data are presented as fold change compared with WT ECs, mean ± SEM. *P < 0.05.
To determine whether individual components of the cluster can regulate FZD4 and LRP6 mRNA levels, we transfected 17∼92 KO ECs with an miR mimic (miR-17a, miR-18a, miR-19a, or miR-92a). Expression of miR-17a, miR-19a, and miR-92a mimics resulted in robust down-regulation of FZD4 mRNA levels (Fig. 4C), with miR-17a and miR-19a predicting complementary sites in the 3′ UTR of FZD4. LRP6 contains a putative target site in its 3′ UTR for miR-19a/b binding; however, overexpression of the individual mimics resulted in a consistent trend toward reduced levels of LRP6 mRNA by miR-18a, but not by miR-19a (Fig. 4C). These results imply that several miRNAs encoded in the miR-17∼92 cluster can cotarget and repress WNT related receptors, either directly or indirectly, and thus have the potential to adjust the plasticity of the WNT signaling pathway in ECs.
We next tested whether miRNAs of this cluster directly target FZD4 and LRP6. To do so, we transfected HEK293T cells with the full-length 3′ UTR of FZD4 or LRP6 cloned into a bicistronic Renilla/firefly luciferase reporter vector and cotransfected with individual mimics or a negative control scramble mimic. miR-19a, but not the other miRNA mimics, reduced the levels of the FZD4 and LRP6 3′ UTR reporter, demonstrating that both of these receptors are targets of miR-19 (Fig. 4D). Mutagenesis of the seed sequences of the predicted miR-19 binding sites restored luciferase expression, confirming the specificity of the interaction between miR-19 and the FZD4 and LRP6 3′ UTR (Fig. S5B). Given that loss of the cluster increased LRP6 protein levels and that miR-19a reduced luciferase activity of the 3′ UTR construct in a sequence-specific manner but did not directly reduce LRP6 mRNA levels, it is likely that miR-19 regulates the translational efficiency of LRP6.
miR-17∼92 Antagonizes the WNT Signaling Cascade.
To investigate the role of the miR-17∼92 cluster on the canonical WNT/β-catenin pathway, we examined WNT3a signaling in WT and miR17∼92 KO ECs. WNT3a treatment increased β-catenin stabilization in cytosolic (Fig. 4E) and nuclear (Fig. 4F) extracts from both EC groups; however, basal and WNT3a-stimulated accumulation of β-catenin was enhanced in the 17∼92 KO ECs. To examine the in vivo relevance of WNT activation in mice lacking the miR-17∼92 in ECs, we crossed BAT-gal reporter mice for WNT/β-catenin signaling (32) to VE-Cad-CreERT2;17∼92fl/fl (17∼92 EC KOVE-Cad) mice. Nuclear WNT reporter gene expression was enhanced in ECs lining small mesenteric arteries of 17∼92 KO mice (17∼92 EC KOVE-Cad/BAT-gal), whereas control mice harboring the BAT-gal reporter and the floxed allele lacking Cre (WT/BAT-gal) did not demonstrate positive staining (Fig. 4G).
Finally, because miR-19a reduces the levels of both FZD4 and LRP6, we examined the effect of miR-19a on WNT/β-catenin–mediated gene expression in ECs. Mouse lung ECs (MLECs) were transfected with either miR-19a mimic or scrambled mimic control. After 48 h, the cells were treated with WNT3a. Transfection with miR-19a cells resulted in reduced expression of several β-catenin–dependent genes in response to WNT3a treatment, including Axin2, Sox17, and Cyclin D1 (Fig. 4H). In addition, because FZD4 is a component of both the canonical (β-catenin) and noncanonical (planar cell polarity) pathways, we examined FZD4 coupling to c-Jun NH2-terminal kinase (JNK). Treatment of ECs with anti–miR-19 enhanced WNT3a stimulation of p-JNK (Fig. S6), implying that miR-19a can also negatively regulate planar cell polarity signaling. Collectively, these data show that the miR-17∼92 cluster, particularly miR-19a, negatively regulates WNT signaling and in turn regulates aspects of arterial development.
Fig. S6.

Anti–miR-19 augments noncanonical WNT stimulation of JNK. MLECs were transfected with control or anti–miR-19 (60 nM each) for 48 h before Wnt3a stimulation. Cells were starved for 4 h, then treated with Wnt3a-conditioned medium for the indicated times. Lysates were collected, run on SDS/PAGE gel, and immunoblotted for p-JNK, total JNK, and Hsp90.
FZD4 Knockdown and miR-17∼92 Cluster Components Rescue Augmented Cord Formation In Cluster-Deficient ECs, and Anti-miR-19 Improves HLI and Up-Regulates Tissue FZD4 and Lrp6.
To examine the contribution of augmented WNT signaling to enhanced EC cord formation in miR-17∼92 KO ECs, we silenced FZD4 via siRNA. FZD4 knockdown reduced cord formation in miR-17∼92 KO ECs (Fig. 5A and Fig. S7), as did treatment of miR-17∼92 KO ECs with the LRP6 inhibitor, dickkopf WNT signaling pathway inhibitor 1 (Dkk1; Fig. S8), demonstrating that accelerated cord formation in cells lacking the cluster is dependent in part on an miR-17∼92–FZD4/Lrp6 axis. To investigate which cluster components, particularly miR-19a, play a role in EC morphogenesis, we conducted rescue experiments by transfecting 17∼92 KO ECs with cluster mimics. miR-17a, miR-18a, and miR-19a, but not miR-92a, mimics inhibited cord formation in 17∼92 KO cells (Fig. 5B). This finding is consistent with previously published data using a spheroid-based assay in ECs, suggesting cell-intrinsic antiangiogenic activity for components of the cluster in ECs (16). The extent of cord formation with the individual miRNAs mimics slightly exceeded that of control WT cells, suggesting that it is the repressive effect of multiple miRNAs from the cluster that integrates the sum of the physiological outcomes.
Fig. 5.

FZD4 knockdown and miR-17∼92 cluster components rescue augmented cord formation in cluster-deficient ECs, and LNA–miR-19 improves flow recovery. (A) The 17∼92 KO ECs transfected with siFZD4 (20 nM) or control siRNA were plated on Matrigel to assess cord formation. *P < 0.05. (B) Individual components of the cluster were transfected into 17∼92 KO ECs, and cord formation was quantified. *P < 0.05. n = 3 experiments in triplicate. (Scale bar: 200 μm.) (C and D) Aged BAT-gal or WT mice were injected with 12.5 mg/kg LNA–anti–miR-19 or LNA scramble control for 3 consecutive days before induction of HLI, followed by injections on days 1, 8, and 15 post-HLI. (C) β-catenin–dependent gene expression (LacZ) in ischemic limbs. (D) Blood flow changes; n = 9 mice per group. *P < 0.05, two way ANOVA. (E) qRT-PCR analysis of thigh muscle tissue confirming miR-19 repression in animals treated with the LNA-modified anti–miR-19. Sample analyses depict overall significant increases in FZD4 (Middle) and LRP6 (Right) mRNA levels in mice treated with LNA miR-19 expression compared with control LNA-miR–treated mice. n = 4 mice per group. *P < 0.05, two-way ANOVA. All data are mean ± SEM.
Fig. S7.

Efficient deletion of FZD4 in 17∼92 KO ECs. The 17∼92 KO ECs were transfected with 10 nM or 30 nM siFZD4. After 48 h, cells were harvested, and mRNA levels of FZD4 were measured by qRT-PCR. The data are representative of an experiment repeated three times and conducted in triplicate. *P < 0.05.
Fig. S8.

DKK1 antagonizes cord formation in ECs from miR-17–92 KO mice. WT and miR 17∼92 KO ECs were placed into cord assays in the presence of 0.5% serum and untreated or treated with Dkk1 (1 mg/mL). Cord formation was monitored over 5–8 h. Data are representative of an experiment conducted in triplicate and subsequently repeated, showing that DKK1 reduces cord formation in KO ECs in a dose-dependent manner. Data are presented as mean ± SEM. *P < 0.05.
Given that miR-19a was found to affect FZD4/LRP6 levels, reduce WNT/β-catenin–dependent gene expression, and rescue aberrant cord formation, we assessed the physiological role of miR-19a in vivo using an LNA–anti-miR approach. Initially, aged BAT-gal mice were subjected to HLI and treated with s.c. injections (3 d before and 2 d after surgery) of an LNA–anti-miR targeting miR19a/b (33), and β-gal expression was measured in tissue. As shown in Fig. 5C, anti–miR-19, but not control, treatment increased reporter gene expression in capillary ECs surrounding regenerating muscle fibers in ischemic tissue. We then performed similar experiments in aged C57Bl6 mice and assessed blood flow recovery over a 4-wk period. Anti–miR-19 increased blood flow recovery compared with control anti-miR–treated mice (Fig. 5C) in a manner comparable to that seen with anti–miR-92a (14). Anti–miR-19 treatment reduced levels of miR-19a in both ischemic and nonischemic tissues (Fig. 5D) and enhanced FZD4 and LRP6 mRNA levels in tissues compared with control treated mice (Fig. 5D). Thus, suppression of miR-19a/b activity regulates arteriogenesis and blood flow recovery, in part through enhanced FZD4 and LRP6 levels.
Discussion
Here we have demonstrated a critical role for the endothelial-derived miR-17∼92 cluster in shaping physiological and ischemia triggered arteriogenesis. The enhanced arterial density in limbs and hearts of mice lacking the cluster points to a developmental role of these miRNAs, as well as an adaptive role in promoting arteriogenesis and function postischemia. Individual cluster components positively or negatively regulate EC functions in vitro, and, remarkably, ECs lacking the cluster spontaneously form cords in a manner rescued by miR-17a, -18a, and -19a or by knockdown of the WNT receptor FZD4. miR-19a negatively regulates FZD4 and its coreceptor LRP6, and β-catenin–dependent gene expression and antagonism of miR-19a/b in aged mice improves blood flow recovery after ischemia and reduces repression of these targets. Collectively, these findings provide novel insights into miRNA regulation of arterialization and highlight the importance of vascular WNT signaling in maintaining arterial blood flow (25).
A key finding of this study is that EC-specific deletion of miR-17∼92 resulted in increased arterial vasculature density in ischemic limbs and consequently improved blood flow recovery compared with control mice. The increased arterial density detected by unbiased 3D micro-CT was threefold in the upper limbs of WT mice and fivefold in miR-17∼92 KO mice. The increased arterial density and improved ischemic recovery in miR-17∼92 KO mice (using two different CRE lines expressed in ECs as well as in a subset of hematopoietic cells), along with the improved blood flow in LNA-miR19–treated aged mice, complement previously reported data showing that antagonism of miR-92a results in enhanced blood flow recovery and blood vessel growth (14). Thus, at least two components of the cluster have evolved to regulate the robustness of arteriogenesis. This occurs despite the fact that miR-19a/b and miR-92a target different spectra of genes and processes, not sharing seed sequence similarity, and exert different actions on transcript levels that overall complement one another functionally. For example, miR-19, but not miR-92a, drives inhibition of cord formation in primary EC cultures, whereas miR-92a regulates integrin alpha-5 expression levels and adhesion (14). Using both in vitro and in vivo analysis, we identified FZD4 and LRP6 as novel targets of miR-19a/b. Both targets were up-regulated in 17∼92 KO ECs compared with control ECs, and both were demonstrated to be targeted by miR-19 using luciferase assays.
Multiple growth-signaling systems are responsible for the growth, maintenance, development, and pathological states of arteriogenesis. These include VEGF, Notch, FGF, angiopoietins, and ephrin ligands that activate canonical signaling pathways leading to an integrated physiological response. Emerging evidence also implicates the WNT/β-catenin pathway in angiogenesis and arteriogenesis. In endothelium, β-catenin–dependent gene expression is essential for balancing Notch-induced vessel stability via Dll4 (20) or, alternatively, via Nrarp (a Notch-regulated gene), which differentially modulates Notch and WNT signaling activity to balance tip and stalk cell proliferation during network formation (20, 34). Of clinical relevance, components of this pathway, FZD4 and LRP5/6, have been associated with inherited vascular diseases, Norrie disease, and familial exudative viteoretinopathy, which manifests in most cases with blindness at birth, persistence of fetal vasculature, and progressive hearing loss (21, 23). Further insights into the contribution of FZD4 and its coreceptor have come from analysis of conditional EC deletion of FZD4 or global FZD4 KO mouse models. Global FZD4 KO mice display a striking reduction in small arterial networks in such tissues as the heart and kidney and impaired blood flow recovery after limb ischemia (25), implying that WNT signaling via FZD4 is critical for arteriogenesis. EC deletion of FZD4 causes hypovascularization in different beds, associated with reduced Sox17 expression (21, 25). These results are opposite of what we observed in EC-specific miR-17∼92–deficient mice and LNA–miR-19–treated mice. In vitro studies have demonstrated the reduced ability of FZD4 KO ECs to form cord-like structures in a Matrigel assay (21, 25). Again, this is in contrast to ECs isolated from miR-17∼92–deficient mice , in which spontaneous cords were formed in a FZD4-dependent manner. Thus, miR-19a/b can repress FZD4/LRP6 signaling and β-catenin/TCF/LEF-dependent gene expression, illustrating the important role of miRNAs in fine-tuning gene expression in the pathways necessary for arteriogenesis.
Based on our results, we propose that miR17∼92 functions as a negative regulator of the WNT/β-catenin pathway. Our working model is that stimuli such as VEGF (3) or fluid shear stress (13) can induce miR-17∼92 cluster expression (4). miR-19 or other components of the cluster, such as miR-18, can repress β-catenin expression (35) or FZD4/LRP6 to attenuate the magnitude or duration of activation of this pathway. Thus, the cluster may provide a feedback mechanism to buffer excessive/leaky β-catenin–dependent transcription. Such feedback loops might be especially relevant and critical to adaptive physiological responses such as arteriogenesis, where the regulation and maintenance of vascular structure and function is critical for blood flow to dependent tissues. Indeed, LNA–miR-19 up-regulation of FZD4/LRP6 levels concomitant with improved ischemic recovery in aged mice supports the dynamic and targetable nature of this response, providing an opportunity to stimulate arteriogenesis.
Materials and Methods
Mice and Crossings.
All animal studies were approved by the Institutional Animal Care and Use Committee of Yale University. Details of genotypes are provided in SI Materials and Methods.
HLI Model and Micro-CT Imaging.
We used 6-mo-old male mice for all experiments, because they have less capacity to completely recover post-HLI. HLI was performed as described previously (28). Total RNA from the semimembranosus and gastrocnemius muscles was isolated using the miRNeasy Mini Kit (Qiagen). A total of 800 ng of RNA was used for cDNA synthesis.
For LNA-miR-19 injection, 6-mo-old male mice (C57Bl6) were injected s.c. with 12.5 mg/kg of LNA-modified anti-miR (miRagen Therapeutics) designed to complement both miR-19a and miR-19b or a universal control. The mice were injected on 3 consecutive days and then subjected to HLI, followed by a weekly maintenance injection throughout the duration of the experiment. The anti–miR-19 belongs to a class of oligonucleotides with a classic LNA-containing oligonucleotide pharmacokinetic profile (36, 37). In brief, plasma concentrations of these anti-miRs typically achieve peak levels between 30 min and 1 h after s.c. administration. Plasma clearance is biphasic, with a short initial distribution phase followed by a longer elimination phase. Oligonucleotide accumulation is greatest in the kidney and liver, with significant accumulation also observed in spleen, bone marrow, and distal skin (away from the injection site). Terminal elimination half-life ranges from roughly 3 to 6 wk.
MLEC Isolation, Cell Culture, and Functional Assays.
Isolation, culture, and functional assays are described in SI Materials and Methods.
Gene Expression Analysis and qRT-PCR.
The analyses are described in SI Materials and Methods.
Immunofluorescence and Immunohistochemistry.
For BAT-gal en face staining of mouse tissue, mesenteric arteries and skeletal muscle were collected from adult mice. Samples were fixed in 4% PFA for 15 min at room temperature and blocked in TNB-T buffer (0.1 M Tris pH 7.4, 150 mM NaCl, 0.2% Triton, 0.5% PerkinElmer blocking reagent) overnight at 4 °C. Samples were stained with BAT-gal antibody (Immunology Consultants Laboratory; RGAL-45A-Z) and PECAM-1 (Millipore; 1398-Z) for 48 h at 4 °C, washed with PBS, and incubated with fluorescent-conjugated secondary antibodies for 2 h at room temperature. Then the samples were washed with PBS, stained with DAPI, fixed with 4% PFA for an additional 10 min, and mounted.
Statistical Analysis.
Statistical analyses were performed using GraphPad Prism 5 software. Significance was tested using the two-tailed unpaired Student t test or two-way ANOVA with Bonferroni correction for multiple comparisons when appropriate. Genome array datasets were analyzed using the paired Student t test. A P value < 0.05 was considered statistically significant. All values are expressed as mean ± SEM.
SI Materials and Methods
Mice and Crossings.
The EC-specific miR-17∼92 KO mice were generated by crossing 17∼92fl/fl females (Mirc1tm1.1TyJ/J) (5) with Tie2-Cre transgenic males [B6.Cg-Tg(Tek-cre)12Flv/J] (26) to generate 17∼92fl/fl (WT) and Tie2-Cre;17∼92fl/fl animals (17∼92 EC KOTie2). Alternatively, endothelial-specific deletion of the cluster was obtained by breeding 17∼92fl/fl females with inducible VE-Cad-Cre-ERT2 transgenic males [Tg(Cdh5-cre/ERT2)1Rha] (17∼92 EC KOVE-Cad) (27). In the latter set, at age 4 mo, Cre activity was induced by five consecutive i.p. injections of tamoxifen (Sigma-Aldrich; T5648) at 0.1 mg/g body weight in a 20-mg/mL stock solution as described previously (36). The mice were administered a second identical regimen at 2 wk before the vascular challenge. To generate mice with WNT reporter gene activity, we mated transgenic 17∼92 EC KOVE-Cad mice with BAT-gal transgenic mice [B6.Cg-Tg(BAT-lacz)3Picc/J] (32) to obtain 17∼92 EC KOVE-Cad; BAT-gal+/− mice. EC-specific KO mice and their WT littermate controls of the same sex were used in this study, and the genetic backgrounds were mixed.
MLEC Isolation, Cell Culture, and Functional Assays.
MLECs were isolated from 5- to 7-wk-old 17∼92 EC KOTie2 or WT mice. All in vitro experiments involving ECs were done with primary EC cultures using cells between passage 0 and passage 3. For signaling analysis of primary cells or immortalized ECs, cells were serum-starved in 0.5% FBS overnight or for 3 h, followed by stimulation with control or 10% WNT3a-conditioned medium for the indicated time points. WNT3a-conditioned and control conditioned media were prepared and tested as described previously using L WNT3a cells (American Type Culture Collection; ATCC CRL-2647) and L control cells (American Type Culture Collection; ATCC CRL-2648). siFZD4 experiments were carried out following standard protocols, and 17∼92 KO ECs were transfected with a final concentration of 10–30 nM siFZD4 as indicated (Ambion). In experiments using miRIDIAN miRNA mimics or control nontargeting mimics, transfection was carried out with 30 nM or 60 nM as indicated (Thermo Scientific Dharmacon).
Gene Expression Analysis and qRT-PCR.
Qiagen Custom RT2 Profiler PCR arrays were used to examine the expression patterns of genes previously validated as targets of cluster components. We compared the gene expression of control and 17∼92 KO primary lung ECs under confluent and optimal growing conditions. Cells were serum-starved for 4 h, plated on a gelatin-coated plate, and collected at 24 h after plating. Total RNA and miRNA-enriched RNA were extracted with the miRNeasy Mini Kit (Qiagen). For the PCR array, reverse transcription was carried out using the RT2 First-Strand Kit (Qiagen), and the array was performed in accordance with the manufacturer’s instructions using the specific RT2 SYBR Green Rox qPCR Master Mix (Qiagen). Real-time PCR amplification reactions were performed on an Applied Biosystems 7900HT Fast Real-Time PCR system. For target modulation in additional experiments, cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad), followed by quantitative analysis using iQ SYBR Green Supermix (Bio-Rad). Primer sequences were as follows: mGAPDH: forward, 5′-AATGTGTCCGTCGTGGATCTGA; reverse, 5-AGTGTAGCCCAAGATGCCCTTC; mFZD4: forward, 5′-AGAGAGAAGAGGGGGAATGG; reverse, 5′-TGTGTGTGGGCTGAAGTGTT; mLrp6: forward, 5′- TGTGGTAAACCCCGAGAAAG; reverse, 5-ATCCTGTTGGCACCTGAGA. GAPDH served as an internal normalization control.
For miRNA detection, extracted RNA was retrotranscribed using the RT2 miRNA First-Strand Kit (Qiagen), and qRT-PCR was performed using SYBR Green Mastermix (Qiagen). Snord66 served as an internal normalization control. For the detection of pri- miRNA, RNA was reverse-transcribed using the High-Capacity RNA to cDNA Kit (Applied Biosystems), and qRT-PCR was performed using Taqman Expression Master Mix (Applied Biosystems). GAPDH served as an internal normalization control. RNA was extracted from three to five biological replicates. Real-time PCR amplification reactions were performed on the iQ5 Real-Time PCR Detection System (Bio-Rad). Data were analyzed using the comparative CT method (also known as the 2−ΔΔCT method).
Proliferation, Cord Assay, and Adhesion Assay.
For the cord formation assay, 48-well plates were coated with 180 μL of Matrigel (BD Biosciences; 354230). Cells were serum-starved with 0.5% FBS for 3 h and then plated at 4 × 104 cells/well in 0.5% FBS alone or supplemented with bFGF at 10 ng/mL (Invitrogen). Cords were imaged at 6–8 h after plating with an Olympus IX 51 microscope. For BrdU proliferation assays, serum-starved cells were seeded in a 96-well plate and treated with 0.5% FBS or 20% (vol/vol) FBS for 24 h. During the last 6 h of treatment, cells were incubated in the presence of BrdU, and its incorporation was detected using Cell Proliferation ELISA, BrdU (Colorimetric) (Roche) according to the manufacturer’s instructions. For adhesion assays, 96-well plates were either uncoated or coated with 0.1% gelatin (Sigma-Aldrich) or fibronectin at 1 μg/cm2 (BD Biosciences) for 3 h at 37 °C. Cells were seeded in a fixed volume of 100 μL. At 30 min after plating, cells were washed with PBS and fixed in 4% (wt/vol) PFA (Electron Microscopy Sciences) in 4 °C for 30 min. Adhesive cells were stained with 0.5% filtered Crystal Violet solution (Sigma-Aldrich) for 1 h at room temperature. Staining solution was aspirated, and cells were rinsed with PBS and allowed to dry. Cells were measured spectrophotometrically at 630 nm. Data are presented as normalized adhesion = adhesion OD value –average uncoated OD adhesion value. Triplicates of each condition were performed in each of the two independent biological experiments.
Endothelial Wound Assay.
Primary MLECs were isolated from miR17-92iCdh5-CRE-ERT2 mice at 6 d after tamoxifen injection. WT and 17∼92 KOVE-Cad ECs were seeded in six-well plates, allowed to reach confluence, and then starved for 4 h in DMEM plus 0.5% FBS. Each well was marked below the plate surface to create landmarks to guide wounding and subsequent image analysis in the same location over various time points. Cells in each well were then removed in four areas using a 200-μL sterile pipette tip. Images of wounded areas were taken at time 0 and 12 h after stimulation with 1 0ng/mL bFGF2. Wounded areas were measured at time 0 (At0) and at time 12 (At12) using ImageJ. Migration was expressed as the percentage of scratch closure after 12 h compared with the corresponding time 0 measurement according to the following formula: % closure = [(At0 − At12)/At0] × 100. Migration in each well was defined as the mean closure of four different scratches. Experiments were performed in duplicate wells, and the results are the combination of two independent experiments.
Aortic Sprouting Model.
The aortic ring microvessel formation assay was performed as reported previously (38). In brief, thoracic aortas were dissected from miR17-92iCdh5-CRE-ERT2 mice at 6 d after tamoxifen injection. Under sterile conditions, aortas were cleaned, transferred to 10-cm culture dishes, and cut into 0.5-mm-thick rings with a sterile scalpel. Rings were then starved with 10 mL of serum-free Gibco Opti-MEM containing GlutaMax (Thermo Fisher Scientific; 51985) plus penicillin (100 U/mL)/streptomycin (100 µg/mL) (Thermo Fisher Scientific; 15140) and then incubated overnight at 37 °C and 5% (vol/vol) CO2. The next day, one ring per mouse was transferred into one well of a 96-well plate containing 50 µL of a freshly prepared solution of collagen type I (Millipore; 08-115). The collagen solution and polymerization protocol for ring embedding were as described previously. Embedded rings were maintained in Opti-MEM + 2.5% (vol/vol) FBS with penicillin-streptomycin, with or without VEGF (100 ng/mL) (R&D Systems; 293-VE-010). The medium was replaced every 2 d for each ring.
On day 6, rings were fixed with PFA [4% (wt/vol)], stained with BS1 Lectin-FITC (1 mg/mL, L9381; Sigma-Aldrich), and imaged with a Nikon fluorescence microscope at 4× magnification. From the images captured, the number of sprouts was counted for each ring, and the sprout numbers per ring were averaged for each group and graphed. The bars in the graphs represent analysis of eight or nine rings per mouse from five or six mice per group.
Luciferase Reporter Assay.
A 1-kb fragment of mouse FZD4 3′ UTR or a 400-bp fragment of mouse LRP6 3′ UTR was generated by qRT-PCR and cloned into the psiCHECK-2 Vector (Promega). HEK293 cells were plated onto a 24-well plate at 9 × 104 cells/well at 24 h before transfection. Then 50 ng of psiCHECK-2 reporter plasmid containing the cloned 3′ UTR or its mutant and 60 nM miRNA mimic (Thermo Scientific Dharmacon) were cotransfected using Lipofectamine 2000 (Invitrogen) and Oligofectamine (Invitrogen), respectively, in triplicate wells. Luciferase assays were performed at 48 h after transfection using the Promega Dual-Luciferase Reporter Assay System.
Western Blot and Nuclear Extraction Analysis.
Immunoblots for cell lysates were performed from whole-cell lysates as described or from nuclear/cytoplasmic fractionation (BioVision). Antibodies used included HSP90 (BD Biosciences; 610419), β-catenin (BD Biosciences; 610154), GAPDH (R&D Systems; AF5718), and fluorescent-labeled antibodies (LI-COR Biotechnologies).
Acknowledgments
This work was supported by National Institutes of Health Grants R01 HL64793, R01 HL61371, R01 HL081190, and P01 HL1070295 and the Leducq Fondation (MicroRNA-Based Therapeutic Strategies in Vascular Disease Network) (to W.C.S.); National Institutes of Health Grants IH R01HL126933 and R56HL117064 and American Heart Association Grant 14GRNT2045009 (to J.Y.); Grant F32-HL107078-02 (to S.L.-E.); and a Leslie H. Warner Postdoctoral Fellowship from the Yale Cancer Center (to S.L.-E.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1507094112/-/DCSupplemental.
References
- 1.Bartel DP. MicroRNAs: Target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiao C, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9(4):405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suárez Y, et al. Dicer-dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc Natl Acad Sci USA. 2008;105(37):14082–14087. doi: 10.1073/pnas.0804597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dews M, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38(9):1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ventura A, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132(5):875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ota A, et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64(9):3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- 7.Mavrakis KJ, et al. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL) Nat Genet. 2011;43(7):673–678. doi: 10.1038/ng.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mestdagh P, et al. The miR-17-92 microRNA cluster regulates multiple components of the TGF-β pathway in neuroblastoma. Mol Cell. 2010;40(5):762–773. doi: 10.1016/j.molcel.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mu P, et al. Genetic dissection of the miR-17∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 2009;23(24):2806–2811. doi: 10.1101/gad.1872909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc–regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 11.Dews M, et al. The myc-miR-17∼92 axis blunts TGFbeta signaling and production of multiple TGFbeta-dependent antiangiogenic factors. Cancer Res. 2010;70(20):8233–8246. doi: 10.1158/0008-5472.CAN-10-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ning G, Liu X, Dai M, Meng A, Wang Q. MicroRNA-92a upholds Bmp signaling by targeting noggin3 during pharyngeal cartilage formation. Dev Cell. 2013;24(3):283–295. doi: 10.1016/j.devcel.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 13.Qin X, et al. MicroRNA-19a mediates the suppressive effect of laminar flow on cyclin D1 expression in human umbilical vein endothelial cells. Proc Natl Acad Sci USA. 2010;107(7):3240–3244. doi: 10.1073/pnas.0914882107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonauer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324(5935):1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 15.Fang Y, Davies PF. Site-specific microRNA-92a regulation of Kruppel-like factors 4 and 2 in atherosusceptible endothelium. Arterioscler Thromb Vasc Biol. 2012;32(4):979–987. doi: 10.1161/ATVBAHA.111.244053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doebele C, et al. Members of the microRNA-17-92 cluster exhibit a cell-intrinsic antiangiogenic function in endothelial cells. Blood. 2010;115(23):4944–4950. doi: 10.1182/blood-2010-01-264812. [DOI] [PubMed] [Google Scholar]
- 17.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cattelino A, et al. The conditional inactivation of the beta-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J Cell Biol. 2003;162(6):1111–1122. doi: 10.1083/jcb.200212157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liebner S, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008;183(3):409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corada M, et al. The Wnt/beta-catenin pathway modulates vascular remodeling and specification by up-regulating Dll4/Notch signaling. Dev Cell. 2010;18(6):938–949. doi: 10.1016/j.devcel.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye X, et al. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009;139(2):285–298. doi: 10.1016/j.cell.2009.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robitaille J, et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat Genet. 2002;32(2):326–330. doi: 10.1038/ng957. [DOI] [PubMed] [Google Scholar]
- 23.Xu Q, et al. Vascular development in the retina and inner ear: Control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116(6):883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 24.He X, Semenov M, Tamai K, Zeng X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: Arrows point the way. Development. 2004;131(8):1663–1677. doi: 10.1242/dev.01117. [DOI] [PubMed] [Google Scholar]
- 25.Descamps B, et al. Frizzled 4 regulates arterial network organization through noncanonical Wnt/planar cell polarity signaling. Circ Res. 2012;110(1):47–58. doi: 10.1161/CIRCRESAHA.111.250936. [DOI] [PubMed] [Google Scholar]
- 26.Koni PA, et al. Conditional vascular cell adhesion molecule 1 deletion in mice: Impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193(6):741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benedito R, et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137(6):1124–1135. doi: 10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 28.Yu J, et al. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc Natl Acad Sci USA. 2005;102(31):10999–11004. doi: 10.1073/pnas.0501444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu Y, Thomson JM, Wong HY, Hammond SM, Hogan BL. Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev Biol. 2007;310(2):442–453. doi: 10.1016/j.ydbio.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He L, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435(7043):828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conkrite K, et al. miR-17∼92 cooperates with RB pathway mutations to promote retinoblastoma. Genes Dev. 2011;25(16):1734–1745. doi: 10.1101/gad.17027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maretto S, et al. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci USA. 2003;100(6):3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olive V, et al. miR-19 is a key oncogenic component of mir-17-92. Genes Dev. 2009;23(24):2839–2849. doi: 10.1101/gad.1861409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phng LK, et al. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev Cell. 2009;16(1):70–82. doi: 10.1016/j.devcel.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang H, et al. Quantitatively controlling expression of mir-17?92 determines colon tumor progression in a mouse tumor model. Am J Pathol. 2014;184(5):1355–1368. doi: 10.1016/j.ajpath.2014.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elmén J, et al. Antagonism of microRNA-122 in mice by systemically administered LNA–anti-miR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008;36(4):1153–1162. doi: 10.1093/nar/gkm1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montgomery RL, et al. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation. 2011;124(14):1537–1547. doi: 10.1161/CIRCULATIONAHA.111.030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baker M, et al. Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc. 2012;7(1):89–104. doi: 10.1038/nprot.2011.435. [DOI] [PubMed] [Google Scholar]