

Significance
Voltage-dependent anion channel 2 (VDAC2), like other VDAC proteins, transports solutes across the outer mitochondrial membrane, but it is the only isoform that supports mitochondrial import of cell death-executing proteins, Bak/Bax. To address the underlying mechanism, we created chimeras of VDAC2 and VDAC1 and tested their competency to rescue Bak insertion and apoptosis in VDAC2-deficient cells. Our gain-of-function studies revealed that the middle domain (123–179 aa) is required for rescue. Loss-of-function studies identified two critical residues (T168, D170). Finally, applying these clues to recently solved structures we identified a discrete site on the cytoplasmic side of VDAC2's pore that likely supports Bak insertion and apoptosis.
Keywords: mitochondria, VDAC2, Bak, tBid, apoptosis
Abstract
Voltage-dependent anion channel (VDAC) proteins are major components of the outer mitochondrial membrane. VDAC has three isoforms with >70% sequence similarity and redundant roles in metabolite and ion transport. However, only Vdac2−/− (V2−/−) mice are embryonic lethal, indicating a unique and fundamental function of VDAC2 (V2). Recently, a specific V2 requirement was demonstrated for mitochondrial Bak import and truncated Bid (tBid)-induced apoptosis. To determine the relevant domain(s) of V2 involved, VDAC1 (V1) and V2 chimeric constructs were created and used to rescue V2−/− fibroblasts. Surprisingly, the commonly cited V2-specific N-terminal extension and cysteines were found to be dispensable for Bak import and high tBid sensitivity. In gain-of-function studies, V2 (123–179) was the minimal sequence sufficient to render V1 competent to support Bak insertion. Furthermore, in loss-of-function experiments, T168 and D170 were identified as critical residues. These motifs are conserved in zebrafish V2 (zfV2) that also rescued V2-deficient fibroblasts. Because high-resolution structures of zfV2 and mammalian V1 have become available, we could superimpose these structures and recognized that the critical V2-specific residues help to create a distinctive open “pocket” on the cytoplasmic surface that could facilitate Bak recruitment.
Voltage-dependent anion channel (VDAC) proteins provide the primary pathway for metabolite and ion flux through the outer mitochondrial membrane (OMM) (1, 2). The VDAC has three isoforms in vertebrates, which show >70% sequence similarity and have a broad and overlapping tissue distribution. The three isoforms can substitute for each other in the metabolite and ion flux (2). However, they also have nonredundant roles in cell function. Striking evidence of an isoform-specific role of VDAC2 (V2) is that embryos of homozygous deletion of Vdac2 alleles die during development (3), whereas Vdac1−/− and Vdac3−/− mice are viable and display mild phenotypes (4, 5). Thus, it is of great relevance to determine the specific interactions and the underlying structural arrangements of V2.
Recent evidence suggests that V2 is involved in Ca2+ transport by cardiac mitochondria (6–8), and specifically interacts with several endogenous cytoplasmic and mitochondria-associated proteins, including steroidogenic acute regulatory protein (StAR) (9), glycogen synthase kinase 3 beta (GSK3β) (10), O-GlcNAc transferase (11), ryanodine receptor 2 (8), tubulin (12), viral protein 5, (13, 14), and drugs [erastin (15) and esfevin (7)]. These interactions facilitate mitochondrial targeting, transport, and signaling processes, and, interestingly, they seem to have striking effects on cell survival. However, the most investigated interaction of V2 is with Bak, a proapoptotic Bcl-2 family protein that is central to OMM permeabilization, allowing the release of intermembrane space proteins like cytochrome c (cyto c) and AIF to the cytosol to induce apoptosis (16, 17).
OMM permeabilization can occur when death receptors like TNF-α receptors are engaged and trigger caspase-8 activation to cleave Bid, a BH3-only proapoptotic Bcl-2 family member. Relocation of truncated Bid (tBid) (18, 19) to the OMM and interaction with Bcl-2 family proteins (either proapoptotic and/or antiapoptotic) lead to a conformational change and subsequent homo-oligomerization or hetero-oligomerization of Bak (19, 20) and another proapoptotic Bcl-2 family protein, Bax (21), providing the means of exit for intermembrane space proteins across the OMM (22–24). At least in several cell types, the rapid kinetics of tBid-induced OMM permeabilization are primarily dependent on Bak (25), presumably because Bak usually resides in the OMM, whereas the mostly cytoplasmic Bax first needs to translocate to the mitochondria (26, 27). Distribution of Bak among individual mitochondria is uneven in cells lacking the OMM fusion proteins Mfn1/2, indicating that OMM fusion is required for Bak distribution (28). However, the availability of Bak in the OMM is primarily controlled by insertion of Bak from cytoplasm to the OMM.
Bak is a tail-anchored protein, but it is different from other tail-anchored proteins like Bcl-2 or Bcl-xL in that unmasking of its C-terminal tail domain and insertion to the mitochondria require V2 (25, 29). In the OMM, Bak was also shown to associate with V2 and to be part of a large complex (3). The interplay between Bak and V2 is central to mitochondrial apoptosis and might contribute to the vital significance of V2 (3, 25, 30–32).
Structural predictions of the VDAC family, primarily VDAC1 (V1) have been done using biochemical methods (33, 34) or biophysical methods (i.e., NMR, crystallography) (35–39). These studies uniformly suggest that VDACs are β-barrel–forming transmembrane channels. Based on the biochemical studies, the β-barrel is composed of one helix and 13 β-sheets with some relatively long loops between β-sheets. In this model, the α-helix is part of the wall of the channel. It had been proposed that the long loops might play a role in protein–protein interaction (1). Differently, however, the biophysical studies show a β-barrel formed by 19 β-sheets with the N-terminal α-helix lying inside the pore. The α-helix is highly flexible and is likely to be associated with VDAC channeling activity for the transfer of metabolites like ATP (40, 41). The literature about the orientation of the VDACs is divergent; however, a recent work by De Pinto and coworkers (42) shows that the C terminus faces the intermembrane space. The only crystallography study of V2 used zebrafish V2 (zfV2) that had >90% sequence similarity to mammalian V2. This work revealed few differences between V1 and V2, mostly confined to the cytoplasmic loops (37).
In the present project, we have studied the domain(s) of V2 that are necessary and/or sufficient for Bak targeting to the OMM and Bak-mediated cyto c release. To this end, the few sequence differences between mammalian V1 and V2 were systematically analyzed by creating a series of chimeric constructs. The functional activity of each construct was tested by evaluating the rescue of Bak’s mitochondrial insertion and tBid-induced OMM permeabilization in Vdac2−/− (V2−/−) mouse embryonic fibroblasts (MEFs). Our results demonstrate that previous suspects like the N-terminal extension and high cysteine (C) content of V2 are irrelevant. Unexpectedly, the sequences of amino acids 168–171 and 123–179 represent the minimum domain of V2, which is necessary and sufficient to provide for Bak import and rapid tBid-induced apoptosis, respectively. In combination with the results of recent structural studies, our findings allow us to propose a mechanism of how the apparently minor sequence differences between V1 and V2 can result in stark functional differences.
Results
Genetic Rescue of V2−/− MEFs Provides an Approach for Studying the Requirements of Mitochondrial Bak Import and Cyto c Release.
We and others have provided evidence that Bak insertion to the OMM and tBid-induced OMM permeabilization are supported only by V2 among VDAC isoforms (25, 29, 30). To establish a paradigm for determining the molecular basis for this nonredundant function of V2, we used V2−/− MEFs and compared their Bak level and their sensitivity to tBid-induced cell death under rescued and nonrescued conditions. First, V2−/− MEFs were infected with V2-adenovirus (avV2), and 36–48 h after the infection, the presence of V2 was confirmed in the cells by immunoblotting membrane lysates using a polyclonal anti-V2 antibody (Fig. 1A). This antibody detects two bands at around 32–34 kDa in MEFs expressing V2; however, only the upper band is relevant because it is the one that is absent in the V2−/− MEFs and is restored in the genetically rescued cells (arrow in Fig. 1A). Immunoblotting also showed Bak in WT cells, which was low in V2−/− cells and was restored upon rescue with avV2. Sensitivity of the cells to tBid was monitored by studying cyto c release from mitochondria to cytosol. Blotting of the membrane and cytosolic fractions against cyto c showed that 25 nM tBid for 5 min fails to release cyto c in V2−/− MEFs but causes release in the rescued cells (Fig. 1B). Digitonin (600 μg/mL) was used as a control to perforate the OMM and mobilize all of the cyto c from mitochondria in both V2-deficient and rescued cells. Because tBid-induced release of cyto c causes mitochondrial membrane potential (ΔΨm) loss in the presence of oligomycin (43), we recorded the ΔΨm in a suspension of permeabilized cells using a fluorescent dye [tetramethylrhodamine methyl ester (TMRM)] (Fig. 1C). In the V2−/− cells (black traces in Fig. 1C), tBid addition did not change the ΔΨm, but in the rescued cells (red traces in Fig. 1C), mitochondria responded to tBid by progressive depolarization. At the end of each experiment, a protonophore [carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP)] was added to dissipate the ΔΨm completely. These results suggest that V2−/− MEFs provide a model allowing mutational analysis of VDAC for Bak recruitment and tBid-induced cyto c release. Furthermore, these findings extend the genetic evidence on the critical role of V2 in mitochondrial Bak import and tBid-induced rapid cyto c release.
Fig. 1.
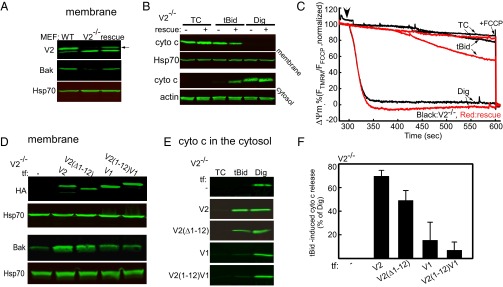
Genetic rescue studies in V2−/− MEFs. Unique N-terminal extension in V2 is neither necessary nor enough for Bak import and cyto c release. (A) Western blot verification of the presence of V2 and Bak in the membrane fraction of WT and V2−/− cells rescued with avV2. The arrow shows the relevant band of V2. (B) Cyto c release was monitored by detecting the cyto c level in the membrane and cytosolic fraction of nonrescued (−) and avV2-rescued (+) V2−/− permeabilized MEFs 5 min after treatment with solvent [tBid (25 nM) or digitonin (Dig, 600 μg/mL)]. TC, time control. Hsp70 and actin were used as loading controls. (C) Representative traces for the time course of ΔΨm recorded in cell suspensions using a fluorometer. The experiment was carried out in permeabilized V2−/− (black line) or avV2-rescued V2−/− (red line) cells treated with tBid or Dig (added at 300 s, marked by an arrow). FCCP (5 μM) was added at the end of the recordings to dissipate the remaining ΔΨm. Data are presented as the percentage of the initial ΔΨm. TCs are the nontreated samples. (D) HA and Bak Western blot of the membrane fraction of V2−/− cells transfected (tf) with HA-tagged V2, V2(Δ1–12), V1, or V2(1–12)V1. Immunoblot (E) and quantitative analysis (F) of cyto c in the cytosolic fraction of V2−/− cells transfected with V2, V2(Δ1–12), V1, or V2(1–12)V1 and treated with tBid or Dig after plasma membrane permeabilization. In response to tBid, the cyto c band was normalized to the response to Dig in each condition (n = 3).
Unique N-Terminal Extension in V2 Is Not Necessary for Bak Import and Cyto c Release.
To identify the motifs of V2 that are necessary and/or sufficient for Bak import and tBid-induced OMM permeabilization, the protein sequence of V2 was systematically compared with V1 and VDAC3 isoforms. The comparison showed a unique 12-aa extension at the N-terminal end of V2 (Fig. S1A), which is conserved in all mammals (Fig. S1B). To test the specific contribution of this domain, the 12-aa extension was either removed in the construct of V2-HA [called V2(Δ1–12)] or added to N terminus of V1-HA [called V2(1–12)V1] (Fig. S2A). V2−/− MEFs were cotransfected with the constructs described above, and mitochondrial matrix-YFP. For negative control the cells were transfected with only mitochondrial matrix-YFP. Forty-eight hours later, cells were sorted on the basis of YFP fluorescence. First, we validated the expression of the mutant VDAC proteins in the sorted V2−/− cells using an anti-HA tag antibody (Fig. 1D, Upper). Second, Bak recruitment was examined by immunoblotting the membrane samples. The Bak level was increased in the V2−/− cells transfected with V2 and V2(Δ1–12), whereas it was similar to negative control in the cells expressing V1 and V2(1–12)V1 (Fig. 1D, Lower).
Fig. S1.

(A) Comparison of the N-terminal segment in mouse VDAC isoforms. (B) Conservation of the N-terminal extension of V2 only in mammals. (C) Cyto c release recorded using dynamics of the cyto c in the V2−/− cells cotransfected with cyto c-GFP and V2, V2(Δ1–12), or V1 in the presence or absence of tBid after 5 min. Data are presented as the changes in fluorescent intensity of GFP normalized to F0. F0 is the intensity of fluorescence at zero second. (D) Representative traces of the time course recording of ΔΨm in single V2−/− cells expressing V2 and Chi12 with tBid (circle) or without (cross). The arrow shows tBid addition. TC, time control. (E) HA and Bak Western blot of V2−/− cells transfected with V2 or M4. tf, transfected.
Fig. S2.
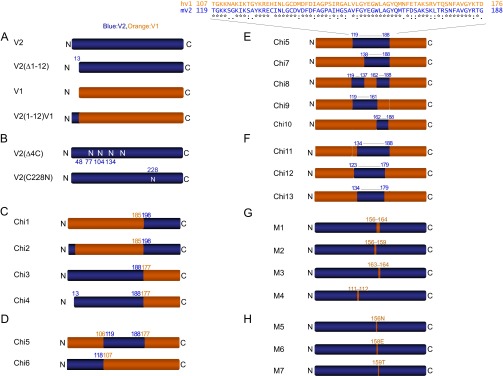
(A–H) Schematic presentation of the constructs used in this study. Blue and orange colors are representative of V2 and V1 sequences, respectively. (B) Location of the replacement of C with N is marked by residue numbers. N228 corresponds to N216 in V1.
Treatment of permeabilized cells with 25 nM tBid for 5 min resulted in cyto c release in the fibroblasts expressing V2 and V2(Δ1–12) but not in the cells expressing V1 or V2(1–12)V1 (Fig. 1 E and F). In addition, cyto c dynamics were monitored in the cells cotransfected with V2, V2(Δ1–12), V1, and cyto c-GFP using fluorescent microscopy. After applying 25 nM tBid, a significant amount of cyto c was rapidly released in cells expressing V2 or V2(Δ1–12) and there was no cyto c release in the cells expressing V1 (Fig. S1C). Altogether, the data confirm that only V2, and not V1, supports Bak targeting and tBid-induced mitochondrial apoptosis effectively. In addition, the data indicate that the 12-aa extension in the N terminus of V2 is not needed to support Bak targeting and tBid-dependent cyto c release.
V2-Specific C’s Are Dispensable for Bak Import and tBid-Dependent OMM Permeabilization.
Comparison of amino acid sequences in V1, V2, and VDAC3 revealed similar values for all residues except C, which is exclusively higher in mammalian V2 (11 vs. 2 in V1 and 6 in V3). Localization of the C’s in the published biophysical model of VDAC (35–38) showed that four of the C’s (48, 77, 104, and 134; shown by blue arrows in Fig. 2A) in V2 are in the linkers between β-sheets. They are seemingly at the same side of the V2 molecule and might be involved in protein–protein interaction (Fig. 2A). To test their possible role in Bak recruitment, the four linker-localized C’s (48, 77, 104, and 134) were mutated to asparagine (N) [called V2(4ΔC)] (Fig. S2B, Upper). Furthermore, based on similarities between structures of V1 and V2 isoforms, we also localized the C’s in the so-called “biochemical model” of V1 (1, 34). In this model, two of the C’s (211 and 228) are in an external loop of V2 (Fig. 2B). We hypothesized that a disulfide bond between neighboring C’s in the loop might stabilize a suitable docking site for Bak or any relevant mediator protein. Therefore, in another mutant, C228 was replaced with its V1-specific counterpart, which is N (N216) (Fig. S2B, Lower). Expression and the presence of the described mutants in the membrane fraction of V2−/− cells expressing V2(4ΔC) and V2(C228N) were confirmed using an anti-V2 antibody (Fig. 2C). V2−/− cells expressing V2(4ΔC) or V2(C228N) showed recruitment of Bak protein to the membrane (Fig. 2C) and were sensitized to tBid (Fig. 2D), indicating that the C’s are not needed for the competence of V2 to support Bak recruitment/tBid-induced cyto c release.
Fig. 2.

V2-specific C’s are dispensable for Bak import and tBid-dependent OMM permeabilization. Schematic view of the mV2 sequence superimposed either with the biophysical model of V1 (38) (A) or the biochemical model (34) (B). The green color shows the β-sheets, whereas red color shows the “not so similar” [scoring >0.5 in the Gonnet point accepted mutation (PAM) 250 matrix] and “different” (scoring ≤0.5 in the Gonnet PAM 250 matrix) amino acids between V1 and V2. Blue arrows show the targeted C locations in V2. (C) V2 and Bak immunoblot of the membrane fractions of the V2−/− cells transfected with V2 (as a positive control), V2(C228N), or V2(Δ4C). (D) Cyto c Western blot for the cytosolic fraction V2−/− cells expressing V2, V2(C228N), or V2(Δ4C) treated with tBid or Dig. V2 and Bak (E, membrane) and cyto c (F, cytosol) Western blots of V2−/− cells expressing mV2 and zfV2.
zfV2 Rescues Bak Import and tBid-Induced OMM Permeabilization in V2−/− MEFs.
To examine further the effect of both the N-terminal extension and the high numbers of C’s in the V2 sequence on the OMM targeting of Bak, we used zfV2 (37). The zfV2 has 82%/92% sequence identity/similarity with mouse V2 (mV2), but it lacks the N-terminal extension and contains only one C at the position of 127. HA-tagged mV2 and zfV2 were transfected in V2−/− cells, and 48 h later, the proteins were detected in the membrane fractions using HA antibody (Fig. 2E). Bak levels in the membrane fraction were similarly elevated by zfV2 and mV2 rescue, indicating that zfV2 is capable of recruiting Bak. Furthermore, treatment of the cells with 25 nM tBid caused similar release of cyto c in the V2−/− cells expressing zfV2 and mV2 (Fig. 2F). Thus, studies of zfV2 further support that the N-terminal extension and C’s are not critical for Bak targeting to the OMM.
Gain-of-Function Studies Using Chimeras of V1 and V2.
As explained above, the biochemical model (Fig. 2B) predicts a long cytoplasmic loop in the C-terminal third of V1 (34), which might be well positioned to support unmasking of the tail of Bak. To find out whether this domain is sufficient or required for Bak targeting, four chimeras were generated. To test whether the loop is sufficient, chimera 1 (Chi1) with the first two-thirds of V1 (1–185) linked to the last third of V2 (198–295) was generated. To test whether the loop is necessary, Chi3 with the first two-thirds of V2 (1–188) linked to the last third of V1 (177–283) was created. Chi2 was created by adding the 12 N-terminal residues to Chi1. Chi4 was created by deletion of this segment from Chi3 (Fig. S2C). V2−/− MEF cells were transfected with the described chimeras, and the presence of relevant proteins was confirmed in the membrane fraction (Fig. 3A). The Bak level did not change in V2−/− cells expressing Chi1 and Chi2, but it increased in the cells expressing Chi3 and Chi4. Furthermore, rescue of tBid-induced cyto c release was apparent only in the cells expressing Chi3 and Chi4 (Fig. 3B). This evidence shows that the last one-third of V2 (198–295), including the long external loop, is neither enough nor required for the tBid/Bak OMM permeabilization pathway. On the other hand, this result indicates that the domain relevant for Bak targeting to the OMM is located between amino acid 12 and amino acid 188 of V2. Therefore, in the next step, we generated two chimeras, Chi5 and Chi6, by substituting one-half of the V2-specific sequence of Chi4 with the relevant sequence of V1 (Fig. S2D). In Chi5, V2 (13–118) of Chi4 was replaced with the relevant sequence of V1, and in Chi6, V2 (119–188) of Chi4 was replaced with the relevant sequence of V1. The presence of the HA-tagged chimeric proteins in the membrane fraction of V2−/− MEF cells expressing Chi5 and Chi6 was confirmed (Fig. 3C). The Bak level increased in the membrane fraction of cells expressing Chi5, but it was unaltered in the cells expressing Chi6 (Fig. 3C). Consistently, tBid released cyto c from V2−/− cells expressing only Chi5 (Fig. 3 D and E). This set of data suggests that the domain that mediates targeting of Bak to OMM is located in the middle domain of V2 (amino acids 119–188).
Fig. 3.
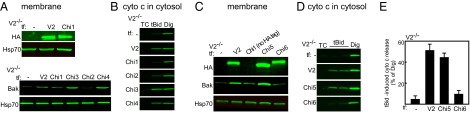
Middle domain of V2 is required for mitochondrial Bak import and tBid-induced cyto c release. (A) HA Western blot (Upper) for the V2−/− cells transfected with HA-tagged V2 or Chi1 and Bak Western blot (Lower) for the cells expressing V2 or Chi1–Chi4. (B) Cyto c Western blot of the V2−/− cells transfected with V2 or Chi1–Chi4. (C) HA and Bak Western blot of the V2−/− cells expressing V2, Chi5, or Chi6. V2−/− expressing nontagged Chi1 was used as a negative control in an HA Western blot. Western blot (D) and quantification (E) of cyto c release in V2−/− cells expressing V2, Chi5, or Chi6 are shown.
V2 (123–179) Is the Shortest Sequence Sufficient for Bak Recruitment.
To narrow the region sufficient for Bak import further, the sequence of V2 in Chi5 was compared with the relevant sequence of V1. Based on clusters of different and not so similar amino acids (using Clustal W nomenclature) in the V2 sequence, we generated three constructs. In each of them, we substituted one-third of the V2-specific middle domain in Chi5 with the corresponding sequence of V1. Specifically, in Chi 7–Chi 9, amino acids 119–137, 138–161, and 162–188 of the V2 sequence were replaced with their V1 counterparts, respectively (Fig. S2E). Expression and proper targeting of the chimeras were confirmed in V2−/− MEF cells expressing Chi 7–Chi 9 (Fig. 4A). In the cells expressing Chi7 and Chi8, the Bak level increased in the membrane fraction, but it did not change in the cells expressing Chi9. The tBid-induced release of cyto c also occurred in the cells expressing Chi7 and Chi8, but no release appeared in the cells expressing Chi9 (Fig. 4B). These data indicate that amino acid residues in the domain of 161–188 are needed for the Bak/cyto c release pathway. To test whether this domain is sufficient for Bak recruitment, Chi10 was generated with V2 (162–188) substituted for the relevant fragment in V1 (Fig. S2E). This mutant was expressed in the V2−/− cells, but it failed to support Bak recruitment (Fig. 4A) and cyto c release after treatment with tBid (Fig. 4B). Thus, V2 (162–188) is essential, but not sufficient, for Bak targeting to the OMM. To determine the N-terminal and C-terminal ends of the region sufficient to substitute for Chi5, new chimeras were generated with various lengths of V2 in Chi5 swapped with the equivalent of V1. In Chi11, the main cluster of differences in the N-terminal end of the V2-specific sequence (119–133) in Chi5 was taken out and was replaced with the corresponding sequence of V1. In Chi12, differences in the C-terminal end of the V2 sequence of Chi5 (180–188) were eliminated by replacement with the relevant part of V1. In Chi13, both V2 (119–133) and V2 (18–188) were cut out and replaced with the relevant fractions of V1 (Fig. S2F). The expression and membrane localization of Chi11–Chi13 were confirmed (Fig. 4C). The Bak level in the membrane fraction and cyto c release were fully restored in the cells expressing Chi12, whereas only partial rescue took place in the cells expressing Chi11 and Chi13 (Fig. 4 C and D). Time lapse monitoring of ΔΨm in adherent cells showed the same pattern of depolarization for V2−/− cells expressing Chi12 or V2 after treatment with tBid (Fig. 4H and Fig. S1D). Altogether, these data show that V2 (123–179) is the shortest sequence that is sufficient for fully supporting the recruitment of Bak and tBid-induced OMM permeabilization.
Fig. 4.

mV2 (123–179) is sufficient and mV2 (156–159) is necessary for Bak import and tBid-induced cyto c release. (A) HA and Bak Western blot of V2−/− cells transfected with Chi7–Chi10 or Chi5. (B) Quantification of cyto c release induced by tBid. (C) Similar to A, except performed for the V2−/− cells expressing Chi5, Chi11–Chi13, or Chi5. (D) Similar to B for Chi11–Chi13 or Chi5-transfected V2−/− cells. (E) Similar to A, except performed for M1–M3. (F) Quantification of cyto c release evoked by tBid for M1–M3. (G) Representative traces of time course recording of ΔΨm in single V2−/− cells expressing M2, M3, or V2 with (thick lines) or without tBid (thin lines). The arrowhead shows tBid addition. FCCP was added to discharge ΔΨm completely at the end of each run. (H) Mitochondrial depolarization presented as the percentage of total ΔΨm in the cells expressing V2, M2, M3, or Chi12 at 7 min after treatment with tBid (n = 3).
Loss-of-Function Mutations in V2: T168 and D170 Are Critical Amino Acids for Bak Targeting to the OMM.
In the next step, we tried to find the critical residues supporting Bak import. Cutting the C-terminal part of V2 in Chi5 (162–188) in Chi9 totally abrogated tBid-induced cyto c release; however, cutting of V2 (180–188) in Chi12 did not change the response. That result led us to hypothesize that the most critical domain must be located in V2 (162–179). The location of this sequence at the edge of a β-sheet (β-sheet 10) in the biophysical model (Fig. 2A) strengthens this idea. In addition, cells expressing Chi7 and Chi13 show a partial response, and that result led us to assume that there might be some effective motifs in the N-terminal part of the V2 sequence in Chi5. Therefore, based on the differences between V2 and V1, which were conserved in all species, we made short mutations in the amino acid 122–179 segment of V2. To test the importance of the C terminus, M1, M2, and M3 were generated in which amino acids 168–176, 168–171, and 175–176 in V2 were replaced with the equivalent sequence of V1 in order. On the other hand, to study the effect of the N-terminal segment, M4 was made by the replacing of V2 (123–124) with the relevant sequence in V1 (Fig. S2G). The presence of mutant proteins in the membrane fraction of V2−/− cells expressing V2 (M1–M4) was validated (Fig. 4E and Fig. S1E). Bak showed an increase in V2−/− cells expressing M3 (Fig. 4E) and M4 (Fig. S1E). By contrast, it did not show any alteration in the cells expressing M1 and M2. The tBid-induced cyto c release was significant in V2−/− cells expressing M3 (similar to the cells rescued by V2). It was moderate in cells expressing M4, and it was abolished in the cells expressing M1 and M2. Also, tBid induced significant depolarization in the V2−/− cells rescued with M3; however, it failed to change ΔΨm in the cells expressing M2 (Fig. 4 G and H). All of the evidence showed that a small sequence of threonine (T), phenylalanine (F), aspartic acid (D), serine (S) or “TFDS” in the position of 168–171 in V2 is critical for Bak insertion and that amino acids serine/glycine (S/G) in the location of 123/124 are also relevant.
In the sequence of TFDS, amino acid F was present both in V1 and V2, so only three amino acids remained to examine. In M5–M7, T, D, and S were replaced with their equivalent in V1, which were asparagine (N), glutamic acid (E), and T (Fig. S2H). The residence of the mutants in the OMM was confirmed in V2−/− cells expressing M5–M7 (Fig. 5A). To check whether each mutant was inserted into the mitochondria, the subcellular distribution of the mutants was analyzed using immunofluorescence staining for the HA tag (red in Fig. 5B). V2−/− cells were cotransfected with GFP-OMP25, an OMM marker (green in Fig. 5B). In all of the conditions, >90% of the red signal overlapped with the area of green signal, which means that the HA-tagged V2 mutant proteins were colocalized with the OMM marker (Fig. 5C). Bak was increased in the V2−/− MEFs expressing M7 (Fig. 5B); however, it was similar to the negative control in cells expressing M5 and M6. Furthermore, tBid rapidly released cyto c and caused collapse of the ΔΨm in cells expressing only M7 (similar to rescued cells with V2) (Fig. 5 D and E). These data demonstrated that two amino acids, including T168 and D170, are the most critical amino acids in mV2-mediated support of the tBid/Bak cell death pathway.
Fig. 5.

Thr167 and Glu169 are the necessary residues in mV2 for mitochondrial Bak import and tBid-induced apoptosis. (A) HA and BAK Western blot of cells expressing M5–M7. (B) Localization analysis of V2 mutants in V2−/− cells coexpressing M5–M7 and GFP-OMP25 (as an OMM marker, green signal) using anti-HA immunofluorescence (red signal). An endoplasmic reticulum (ER)-targeted protein with an HA tag (ER-HA) was used as a negative control. Scale bar on top left image is equal to 5 um. (C) Colocalization of HA immunofluorescence with mitochondria presented as the percentage of the overlap between the green and red areas and the total area of the red signal. (D) Cyto c Western blot of the V2−/− cells expressing V2 or M5–M7. (E) ΔΨm recorded as described for Fig. 4J for the V2−/− cells expressing M5–M7. Arrows show the time of the addition of tBid.
Structural Implications of the Domains Identified by Mutagenesis.
The structure of a mammalian V2 has not been solved. Fortunately, the zfV2 structure has recently become available (37), and our data showed that zfV2, like mV2, supports the Bak/tBid pathway (Fig. 2). The regions corresponding to the sufficient motifs and necessary residues of mV2 are shown in magenta and yellow, respectively in the zfV2 structure [Protein Data Bank (PDB) ID code 4bum] (Fig. 6 A and B). The side view in Fig. 6A shows that T168 (S156 in zfV2) is almost at the end of β-sheet 10 and D170 (D158 in zfV2) is located in the beginning of the loop between β-sheets 10 and 11. The top view shows that both of the necessary residues are on the cytosolic side of the channel and the side chains of both of these amino acids are facing the inner side of the pore. The positioning of these amino acids supports the possibility that the interaction with any other proteins (either directly with Bak or a third party needed for Bak) might be affected from the inner side of the channel. Further along this line, the S171 (T159 in zfV2) that was not critical for Bak recruitment projects to the outer surface.
Fig. 6.

Three-dimensional structure of zfV2. (A and B) Side and top views of zfV2 (PDB ID code 4bum) illustrated using Pymol. Pink is the sufficient segment, and yellow (sticks) is the necessary residues, including S156 and D158. (C, Right) Side view of the segment containing amino acids S156 and D158 (yellow sticks) in zfV2 vs. the relevant residues in mV1 (N156 and E158, orange sticks). Residues 111–167 in zfV2 (blue) and mV1 (PDB ID code: 3emn, orange) were superimposed. (C, Left) Interior view of the channels showing the residues that are within a distance of 4 Å from either S156 or D158 in zfV2 (sticks with carbon colored blue) or N156 or E158 in V1 (sticks with carbon colored orange). In sticks, oxygen is red and nitrogen is green. Adjacent residues have also been marked in the sequences of zfV2 (blue) and mV1 (orange).
Next, the crystal structure of zfV2 (111–168) was aligned with mV1, which cannot support Bak recruitment and has a crystal structure determined at a similar resolution to zfV2 (36). Comparison of the crystal structure of zfV2 with mV1 (PDB ID code 3emn) showed that N156 and E158 in V1 form extensions oriented toward each other to close the gap between them, whereas S156 and D158 diverge to create an open space in zfV2 (Fig. 6C, Upper Right). The presence of S156 and D158 in zfV2 also causes differences in the proximity of adjacent residues (Fig. 6C, Upper Left). In Fig. 6C, sticks represent the amino acids that are within a distance of 4 Å from either S156 or D158 in zfV2 (blue carbons) or either N156 or E158 in V1 (orange carbons). These adjacent amino acids have been indicated in the sequences in Fig. 6C (Lower). Based on the distance between residues in 3D structures, it seems the loops between β-sheets 10 and 11 and between β-sheets 9 and 8 in zfV2 are in closer vicinity than the equivalent loops in mV1. Based on the greater accessibility of the space defined by the V2-specific critical residues and the differential arrangement of their neighbors, it is tempting to assume that V2 might provide a more suitable docking site for Bak recruitment. Notably, the small sequence difference in the highlighted region seems to result in differential plane positioning of the loop between β-sheets 10 and 11 relative to the lipid bilayer (Fig. S3) [predicted by the Orientations of Protein in Membranes (OPM) database (44)], providing another factor for differential isoform-specific interaction with other proteins.
Fig. S3.
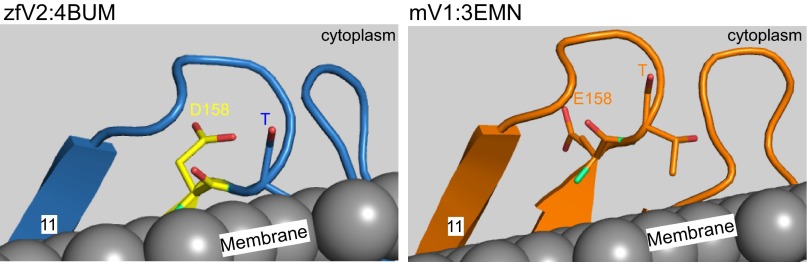
Positioning of the loop between β-sheets 10 and 11 in zfV2 (blue). mV1 has been predicted using the OPM database and illustrated using PyMOL.
Discussion
In this study, we identified by mutational analysis the domain of V2, which plays a role in mitochondrial Bak import and tBid-Bak–mediated apoptosis. Although the N-terminal extension and the higher C content were the suspects previously, we demonstrated that these unique features of the V2 structure are dispensable. Our gain-of-function studies revealed that V2 (123–179) replacement in V1 is sufficient to support the Bak pathway. Furthermore, loss-of-function studies demonstrated that even a small change at T168 or D170 of V2 is sufficient to arrest the Bak pathway and an alteration at S123 or G124 also suppresses this pathway. Based on the available crystal structure, we predict that these residues might help to create a potential docking site for Bak or an adaptor protein. Thus, our findings come together to support an unexpected model for the distinctive, and potentially vital, apoptotic function of V2.
Bak and Bax use their C-terminal α-helix (α9) to target the OMM. Bak is constitutively present in the OMM and mediates the acute phase of Bid-induced OMM permeabilization, whereas Bax, which is predominantly cytosolic in healthy cells, inserts and oligomerizes at the OMM more slowly (25, 45). Differently from other C-terminal anchored proteins, Bak requires V2 for insertion of its hydrophobic C terminus to the OMM (29), and Bax targeting also seems to depend on the presence of either Bak (the hetero-oligomerization partner of Bax) or V2 (30). Furthermore, fusion of the isolated C terminus of Bak to the C terminus of GFP results in mitochondrial targeting of GFP independent of V2. Thus, V2 might interact with a remote site on Bak to unmask α9 that would otherwise be unavailable for insertion to the OMM in the context of the full-length Bak (29, 46). However, how the structure of V2 supports the interaction with Bak and insertion of this protein to the OMM had not been investigated.
A mystery, and a unique opportunity for mutational analysis, was presented by the fact that the three VDAC isoforms display high sequence similarity, but only V2 can facilitate Bak localization to the OMM. Thus, to ensure that the β-barrel channel structure is maintained in the constructs and to avoid protein misfolding, we did not use deletion and created chimeric constructs by V2-specific substitutions in V1 and by V1-specific substitutions in V2. In addition, because of the lack of an antibody that can recognize the chimeras, we decided to use C-terminally HA-tagged constructs. Although a previous work described mitochondrial toxicity, fragmentation, and a drop in ΔΨm when C-terminally modified V1 was expressed and raised concern about C-terminal tagging of β-barrel channels (47), we did not observe any of the above-described problems and, most importantly, we had proper mitochondrial localization and rescue of function with expression of either V2-HA or nontagged V2 in V2−/− MEFs.
We first focused on the most striking sequence differences between V2 vs. V1 and VDAC3, which are the extended N terminus and the relatively high C content of V2. The possible role of these V2-specific features was also suggested by the facts that (i) based on biophysical model, the N terminus is in the pore (35–39, 41) and might interact with cytoplasmic proteins, and (ii) the C’s have been implicated in strengthening the channel interaction with their environment and in stabilization of the channel (48). However, we found that the N-terminal extension and the surplus C’s are dispensable for Bak import and tBid-mediated apoptosis (Fig. 2).
The remaining scattered and subtle sequence differences were evaluated initially with gain-of-function studies, which concluded that 123–179 is the minimum sequence of V2 sufficient to lend competency to V1 to target Bak to the OMM (Figs. 3 and 4). If either the N-terminal or the middle third of this sequence is switched back to V1, the gain of function is partial, whereas reversing back 161–179 to V1 precludes any Bak recruitment and tBid-induced cyto c release (Fig. 4). When the few isoform-specific residues in this sequence were further tested in loss-of-function studies, replacement of V2 (123–124) with the corresponding sequence of V1 caused partial loss of function, whereas replacement of V2 (168–171) led to complete loss of function (Fig. 4). Finally, point mutations at position T168 or D170 of V2 caused essentially complete loss of function (Fig. 5). Thus, multiple residues in the 123–179 sequence contribute to formation of the relevant conformation, and T168 or D170 is an essential residue.
Because this study is, to our knowledge, the first systematic mutational analysis of V2, there is little literature to provide a context for our results. To predict how the essential residues contribute to a potential protein–protein interaction site, we considered the available structures. Although mammalian V2 remains to be studied, the structure of zfV2 has just been solved, and we found that zfV2 rescues Bak import and tBid sensitivity in V2−/− MEF mitochondria. In zfV2, the sufficient sequence spans from 111 to 167 and the essential residues are S156 and D158 (Table 1). As Fig. 6 shows, S156 is almost at the end of β-sheet 10 and D158 is located at the beginning of the loop between β-sheets 10 and 11. This loop is at the cytosolic side of the channel (42), and the side chains of both of these amino acids are facing the inner side of the pore, indicating possible interaction with cytoplasmic proteins (either directly with Bak or with an adaptor) from the inner side of the channel. Consistent with the proposed relevance of the inner surface, mutation of S171 in mV2, the equivalent of T159, whose side chain in mV1 is directed to the outer surface of the channel, did not affect Bak recruitment. Interestingly, S123 and G124 (S111 and G112 in zfV2), the replacement of which by V1-specific residues caused partial loss of Bak-related activity (M4 mutant in Fig. 4), are also at the cytoplasmic end of a β-sheet (β-sheet 7) (Fig. 6A, marked as 111) and so are well positioned to contribute to the formation of a surface domain that supports Bak recruitment.
Table 1.
Alignment of the “sufficient” sequence of V2 from different species and mV1
 |
Because the structures of both zfV2 and mV1 are available at very similar resolutions, we could examine the structural difference caused by the critical residues. Despite the similarity between zfV2 and mV1 in the critical amino acids, comparison of the superimposed structures showed that N156 and E158 in V1 form a closed area, whereas S156 and D158 in zfV2 form the edge of an open “cup.” The wider space created by the side chains of S156 and D158 might favor protein–protein interaction. In addition, this small sequence difference might lead to a change in positioning of the loop between β-sheets 10 and 11 relative to the lipid membrane [as has been predicted by the OPM database (44)], and thereby might affect the affinity for interacting with other proteins. Notably, a recent computational study concluded that A294 in V2 is the primary residue involved in the interaction with Bak (49). However, our experiments provided clearcut evidence that the V2-specific C terminus is not required for rescuing Bak targeting in V2−/− MEF mitochondria (Fig. 3).
In addition to spatial positioning of the residues, posttranslational modification of T168 or D170 might be the reason for the competence of V2 to support mitochondrial Bak import. Indeed, Palaniappan et al. (11) showed that V2 might be a substrate for O-linked β-N-acetylglucosamination; however, our amino acids of interest have not been reported as the location for glycosylation. Furthermore, to study the posttranslational modification of V2, we used tandem MS of V2-HA enriched by immunoprecipitation. Unfortunately, the spectra recorded upon digestion with both trypsin and endoproteinase Asp-N did not reveal our segment of interest. Finally, the possibility of V2 phosphorylation was tested by searching the phosphosite database (50). However, there are no data available for amino acids 132–180 of V2. Lack of data for this part of V2 might be because of a technical difficulty in protein enrichment or because conventional trypsin digestion does not provide fragments covering the sequence of interest. Thus, testing of the possible role of posttranslational modifications needs further refinement of the technology.
Collectively, our results have addressed the question of what V2-specific structural differences allow only this VDAC isoform to control mitochondrial Bak import and, in turn, the acute phase of Bid-mediated apoptosis. Beyond the fatal outcome of murine V2 deletion, the human disease relevance of V2 dysregulation is supported by multiple protein and gene expression datasets showing specific V2 up-regulation in human hepatocarcinomas. Therefore, the structural basis of the V2–Bak interplay might become important in identifying new drug targets. Furthermore, it is of interest to find out whether the V2-specific sequence supporting the Bak pathway is also relevant for the recently noticed interactions of V2 with other proteins, including StAR and GSK3β.
Materials and Methods
Plasmids.
The mV2-HA and human VI (hV1)-HA were cloned into pEYFPN1. The rest of the constructs were generated using relevant restriction enzymes for V2 and V1 sequences (Fig. S4). Cloning was performed using standard molecular biology methods. All of the clones were confirmed with sequencing. All of the constructs have been linked to a chemical tag (human influenza HA), except V2(Δ4C) and V2(C228N). Due to the similarity between hV1 and mV1 (with only four not so different amino acids) and the accessibility of hV1, we used hV1 in our study. Mitochondrial matrix targeting sequence linked to YFP, Mitomatrix-YFP (Clontech) and GFP-OMP25 targeting sequence of OMP25 linked to GFP, GFP-OMP25 (from Manuel Rojo, Université Pierre et Marie Curie, Bordeaux, France) used as mitochondrial markers and cyto c-GFP have been described previously (28).
Fig. S4.



Sequences of the constructs that have been used in this study.
Cell Culture, Transfection, and Electroporation.
MEFs were cultured in DMEM supplemented with 10% (vol/vol) FBS, penicillin and streptomycin, l-glutamine, and nonessential amino acids (Invitrogen). Constructs were introduced to the cells (4.5–6 * 106 cells per 300 μL) by electroporation using BTX-830 square-pulse generator (BTX) (25). Fluorescence-based sorting was performed 48 h after electroporation using 15–20 * 106 cells cotransfected with a VDAC construct or pcDNA and Mitomatrix–YFP or GFP-OMP25. Infection with V2 adenovirus (Vector Biolabs) was done using 200 particles per cell for 36–48 h. For single-cell imaging, 12,000–18,000 cells per well were plated onto glass-bottomed 96-well plates.
ΔΨm and Cyto c Release Assay in Suspensions of Permeabilized Cells.
ΔΨm was measured in suspensions of permeabilized cells using a fluorometer (Delta RAM; PTI) with 540-nm excitation and 580-nm emission for TMRM (2 μM) (25, 51). Briefly, 2.4 mg of cells was resuspended in 1.5 mL of ICM [120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM Hepes/Tris (pH 7.2), supplemented with 1 μg/mL each antipain, leupeptin, and pepstatin] and were permeabilized using digitonin (30–40 μg/mL) for 5 min at 37 °C. Measurements were carried out in the presence of 2 mM MgATP, 2 mM succinate, and 5 μg/mL oligomycin. In case of sorted cells, due to the limitation in cell mass, the experiment was scaled down: 4.5–6 *105 sorted cells were permeabilized in 600 μL of ICM, divided into four equal aliquots used (i) for time control, (ii and iii) for treatment with tBid, and (iv) with digitonin (600 μg/mL) as a positive control. Treatment with recombinant tBid (25 nM) lasted for about 5–7 min. At the end of the incubation period, the cytosolic and membrane fractions were separated by centrifugation at 10,000 × g for 5 min. Pellets were lysed in radioimmunoprecipitation assay buffer, containing 150 mM NaCl, 1.0% (vol/vol) Octylphenoxypolyethoxyethanol (IGEPAL), 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris (pH 8.0; Sigma), and the lysed pellets and cytosol fractions were used for immunoblotting.
Fluorescent Imaging.
Cells were cotransfected with the plasmid of interest and cyto c-GFP, and were preincubated with 2% (wt/vol) BSA containing 25 nM TMRM for 10–15 min at 37 °C. Then, the cells were washed with prewarmed sodium-Hepes-EGTA consisting of 120 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 0.2 mM MgCl2, 20 mM NaOH/Hepes, and 0.1 mM EGTA9 (pH 7.4), and were permeabilized with 40 μg/μL digitonin in ICM at 37 °C. After 5 min, the permeabilization buffer was replaced with fresh ICM (including 5 μg/mL oligomycin, 2 mM succinate, and 2 mM ATP without digitonin). To study cyto c release, redistribution of cyto c-GFP from mitochondria to the cytosol was monitored (51). The imaging was performed at 490/20-nm excitation and 540/50-nm emission using a ProEM1024 EM-CCD (Princeton Instruments), fitted to a Leica DMI 6000B inverted epifluorescence microscope. Confocal imaging experiments were performed using a Zeiss LSM780 system (40× or 20× objective) (28, 51). The laser source was used for imaging of TMRM at 561-nm excitation. The computer-controlled motorized stage allowed us to measure ΔΨm in four different wells simultaneously. During the experiment, 5–8 nM TMRM was used. Also, at the end of each recording, an uncoupler (5 μM FCCP) was added. All analysis was done using customized imaging software.
Immunoblotting.
Western blotting has been described previously (28). Antibodies used include Anti-Bak (no. 06-536; Millipore) Anti-cyt c (no. 556433; BD Bioscience), Anti-HA (no. 9110; Abcam), Anti-V2 (no. ab37985; Abcam), Anti-mtHsp70 (no. MA3-028; Thermo Scientific), and Anti-prohibitin (no. ab28172; Abcam). Detection of bands was performed on a LI-COR Odyssey scanner. ImageJ (NIH) was used for quantification of the bands.
Immunocytochemistry.
Cells cotransfected with the construct of interest and GFP-OMP25 were washed with PBS and then were fixed by prewarmed 3.5% (vol/vol) paraformaldehyde in PBS for 15 min at room temperature. Cells were further permeabilized and blocked with 3% (wt/vol) BSA and 0.1% Tween-20 in PBS for 1 h. They were then incubated for 1 h with Anti-HA antibody in 1% (wt/vol) BSA and 0.1% Tween-20. Finally, the cells were incubated for 45 min with Alexa Fluor 647 anti-rabbit secondary antibody (Molecular Probes).
Acknowledgments
We thank Drs. Jaunian Chen and William Craigen for providing us with zfV2 plasmid and V2−/− MEFs, respectively, and Drs. John Pascal, Suresh Joseph, Soumya Sinha Roy, David Weaver, Gyorgy Csordas, Erin Seifert, and Atan Gross for advice and discussion. This work was supported by NIH Grant GM59419 (to G.H.) and by Hungarian Scientific Research Fund Grant OTKA K105006 (to P.V.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1510574112/-/DCSupplemental.
References
- 1.Colombini M. VDAC: The channel at the interface between mitochondria and the cytosol. Mol Cell Biochem. 2004;256-257(1-2):107–115. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- 2.Craigen WJ, Graham BH. Genetic strategies for dissecting mammalian and Drosophila voltage-dependent anion channel functions. J Bioenerg Biomembr. 2008;40(3):207–212. doi: 10.1007/s10863-008-9146-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301(5632):513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 4.Anflous K, Armstrong DD, Craigen WJ. Altered mitochondrial sensitivity for ADP and maintenance of creatine-stimulated respiration in oxidative striated muscles from VDAC1-deficient mice. J Biol Chem. 2001;276(3):1954–1960. doi: 10.1074/jbc.M006587200. [DOI] [PubMed] [Google Scholar]
- 5.Sampson MJ, et al. Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J Biol Chem. 2001;276(42):39206–39212. doi: 10.1074/jbc.M104724200. [DOI] [PubMed] [Google Scholar]
- 6.Subedi KP, et al. Voltage-dependent anion channel 2 modulates resting Ca²+ sparks, but not action potential-induced Ca²+ signaling in cardiac myocytes. Cell Calcium. 2011;49(2):136–143. doi: 10.1016/j.ceca.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Shimizu H, et al. Mitochondrial Ca(2+) uptake by the voltage-dependent anion channel 2 regulates cardiac rhythmicity. eLife. 2015;4:4e0481. doi: 10.7554/eLife.04801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Min CK, et al. Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca²+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem J. 2012;447(3):371–379. doi: 10.1042/BJ20120705. [DOI] [PubMed] [Google Scholar]
- 9.Prasad M, et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)-voltage-dependent anion channel 2 (VDAC2) interaction. J Biol Chem. 2015;290(5):2604–2616. doi: 10.1074/jbc.M114.605808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanno M, et al. Translocation of glycogen synthase kinase-3β (GSK-3β), a trigger of permeability transition, is kinase activity-dependent and mediated by interaction with voltage-dependent anion channel 2 (VDAC2) J Biol Chem. 2014;289(42):29285–29296. doi: 10.1074/jbc.M114.563924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palaniappan KK, et al. A chemical glycoproteomics platform reveals O-GlcNAcylation of mitochondrial voltage-dependent anion channel 2. Cell Reports. 2013;5(2):546–552. doi: 10.1016/j.celrep.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maldonado EN, et al. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: Regulation by free tubulin and erastin. J Biol Chem. 2013;288(17):11920–11929. doi: 10.1074/jbc.M112.433847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin W, et al. The association of receptor of activated protein kinase C 1(RACK1) with infectious bursal disease virus viral protein VP5 and voltage-dependent anion channel 2 (VDAC2) inhibits apoptosis and enhances viral replication. J Biol Chem. 2015;290(13):8500–8510. doi: 10.1074/jbc.M114.585687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, et al. Critical role for voltage-dependent anion channel 2 in infectious bursal disease virus-induced apoptosis in host cells via interaction with VP5. J Virol. 2012;86(3):1328–1338. doi: 10.1128/JVI.06104-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yagoda N, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014;345(6203):1250256. doi: 10.1126/science.1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: Orchestrators of apoptosis. Biochim Biophys Acta. 2011;1813(4):508–520. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 18.Yin XM. Bid, a BH3-only multi-functional molecule, is at the cross road of life and death. Gene. 2006;369:7–19. doi: 10.1016/j.gene.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 19.Wei MC, et al. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korsmeyer SJ, et al. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7(12):1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 21.Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20(3):929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuwana T, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111(3):331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 23.Letai A, et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2(3):183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 24.Willis SN, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315(5813):856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 25.Roy SS, Ehrlich AM, Craigen WJ, Hajnóczky G. VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria. EMBO Rep. 2009;10(12):1341–1347. doi: 10.1038/embor.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schellenberg B, et al. Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol Cell. 2013;49(5):959–971. doi: 10.1016/j.molcel.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem. 1998;273(17):10777–10783. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- 28.Weaver D, et al. Distribution and apoptotic function of outer membrane proteins depend on mitochondrial fusion. Mol Cell. 2014;54(5):870–878. doi: 10.1016/j.molcel.2014.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Setoguchi K, Otera H, Mihara K. Cytosolic factor- and TOM-independent import of C-tail-anchored mitochondrial outer membrane proteins. EMBO J. 2006;25(24):5635–5647. doi: 10.1038/sj.emboj.7601438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma SB, et al. Bax targets mitochondria by distinct mechanisms before or during apoptotic cell death: A requirement for VDAC2 or Bak for efficient Bax apoptotic function. Cell Death Differ. 2014;21(12):1925–1935. doi: 10.1038/cdd.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lazarou M, et al. Inhibition of Bak activation by VDAC2 is dependent on the Bak transmembrane anchor. J Biol Chem. 2010;285(47):36876–36883. doi: 10.1074/jbc.M110.159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren D, et al. The VDAC2-BAK rheostat controls thymocyte survival. Sci Signal. 2009;2(85):ra48. doi: 10.1126/scisignal.2000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blachly-Dyson E, Peng S, Colombini M, Forte M. Selectivity changes in site-directed mutants of the VDAC ion channel: Structural implications. Science. 1990;247(4947):1233–1236. doi: 10.1126/science.1690454. [DOI] [PubMed] [Google Scholar]
- 34.Song J, Midson C, Blachly-Dyson E, Forte M, Colombini M. The topology of VDAC as probed by biotin modification. J Biol Chem. 1998;273(38):24406–24413. doi: 10.1074/jbc.273.38.24406. [DOI] [PubMed] [Google Scholar]
- 35.Bayrhuber M, et al. Structure of the human voltage-dependent anion channel. Proc Natl Acad Sci USA. 2008;105(40):15370–15375. doi: 10.1073/pnas.0808115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ujwal R, et al. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci USA. 2008;105(46):17742–17747. doi: 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schredelseker J, et al. High resolution structure and double electron-electron resonance of the zebrafish voltage-dependent anion channel 2 reveal an oligomeric population. J Biol Chem. 2014;289(18):12566–12577. doi: 10.1074/jbc.M113.497438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hiller S, et al. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science. 2008;321(5893):1206–1210. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gattin Z, et al. Solid-state NMR, electrophysiology and molecular dynamics characterization of human VDAC2. J Biomol NMR. 2014;61(3-4):311–320. doi: 10.1007/s10858-014-9876-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choudhary OP, et al. Structure-guided simulations illuminate the mechanism of ATP transport through VDAC1. Nat Struct Mol Biol. 2014;21(7):626–632. doi: 10.1038/nsmb.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geula S, Ben-Hail D, Shoshan-Barmatz V. Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem J. 2012;444(3):475–485. doi: 10.1042/BJ20112079. [DOI] [PubMed] [Google Scholar]
- 42.Tomasello MF, Guarino F, Reina S, Messina A, De Pinto V. The voltage-dependent anion selective channel 1 (VDAC1) topography in the mitochondrial outer membrane as detected in intact cell. PLoS One. 2013;8(12):e81522. doi: 10.1371/journal.pone.0081522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madesh M, Antonsson B, Srinivasula SM, Alnemri ES, Hajnóczky G. Rapid kinetics of tBid-induced cytochrome c and Smac/DIABLO release and mitochondrial depolarization. J Biol Chem. 2002;277(7):5651–5659. doi: 10.1074/jbc.M108171200. [DOI] [PubMed] [Google Scholar]
- 44.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: Orientations of proteins in membranes database. Bioinformatics. 2006;22(5):623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 45.Sarosiek KA, et al. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell. 2013;51(6):751–765. doi: 10.1016/j.molcel.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shore GC. Apoptosis: It’s BAK to VDAC. EMBO Rep. 2009;10(12):1311–1313. doi: 10.1038/embor.2009.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kozjak-Pavlovic V, Ross K, Götz M, Goosmann C, Rudel T. A tag at the carboxy terminus prevents membrane integration of VDAC1 in mammalian mitochondria. J Mol Biol. 2010;397(1):219–232. doi: 10.1016/j.jmb.2010.01.047. [DOI] [PubMed] [Google Scholar]
- 48.Maurya SR, Mahalakshmi R. Cysteine residues impact the stability and micelle interaction dynamics of the human mitochondrial β-barrel anion channel hVDAC-2. PLoS One. 2014;9(3):e92183. doi: 10.1371/journal.pone.0092183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veresov VG, Davidovskii AI. Structural insights into proapoptotic signaling mediated by MTCH2, VDAC2, TOM40 and TOM22. Cell Signal. 2014;26(2):370–382. doi: 10.1016/j.cellsig.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 50.Hornbeck PV, et al. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40(Database issue):D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia-Perez C, et al. Bid-induced mitochondrial membrane permeabilization waves propagated by local reactive oxygen species (ROS) signaling. Proc Natl Acad Sci USA. 2012;109(12):4497–4502. doi: 10.1073/pnas.1118244109. [DOI] [PMC free article] [PubMed] [Google Scholar]