

Significance
The neuroinvasive herpesviruses cause a wide range of diseases in humans and livestock. These viruses have the unusual ability to routinely enter the mammalian nervous system following infection of mucous membranes. Although infection is often asymptomatic, the consequences of the resulting lifelong nervous system infections are not fully understood. This study uncovers two opposing nervous system protective barriers that the virus defeats by changing its properties during the course of infection. The molecular switch governing this change underlies the herpesvirus neuroinvasive phenotype.
Keywords: herpesvirus, PRV, HSV-1, neuroinvasion, ubiquitin
Abstract
Neuroinvasive herpesviruses display a remarkable propensity to enter the nervous system of healthy individuals in the absence of obvious trauma at the site of inoculation. We document a repurposing of cellular ubiquitin during infection to switch the virus between two invasive states. The states act sequentially to defeat consecutive host barriers of the peripheral nervous system and together promote the potent neuroinvasive phenotype. The first state directs virus access to nerve endings in peripheral tissue, whereas the second delivers virus particles within nerve fibers to the neural ganglia. Mutant viruses locked in either state remain competent to overcome the corresponding barrier but fail to invade the nervous system. The herpesvirus “ubiquitin switch” may explain the unusual ability of these viruses to routinely enter the nervous system and, as a consequence, their prevalence in human and veterinary hosts.
Herpesviruses such as herpes simplex virus type 1 (HSV-1) and pseudorabies virus (PRV) routinely enter the peripheral nervous system following casual contact between infected and uninfected individuals. Recent findings have provided insights into this complex phenotype, but the molecular mechanisms directing nervous system invasion are poorly understood (1–5). A critical effector of this process is a conserved herpesvirus deubiquitinase (DUB) (a cysteine protease that removes ubiquitin from target posttranslationally modified proteins) (6–8). The DUB is housed in the amino terminus of the pUL36 protein (protein encoded by the 36th open reading frame in the unique-long segment of the viral genome), amounting to less than 10% of the total mass of this large (>250 kDa) structural component of the virion (9). pUL36 is attached directly to the icosahedral capsid shell of these enveloped viruses (10–12) and, due to its location between the capsid and envelope, is referred to as a herpesvirus tegument protein. Upon virus entry into a cell, pUL36 becomes exposed to the cytosol (4, 13, 14) and directs delivery of the capsid and its DNA content to the nucleus (2, 15–19). Following replication, pUL36 is also essential for viral assembly and egress (20–22).
The presence of the pUL36 DUB on the exposed surface of the intracellular viral particle suggests that removal of ubiquitin from cellular cytosolic proteins, or from the virus particle itself, is instrumental for these viruses to transmit between peripheral tissues and the neurons that innervate them (8). This necessarily implicates the prior addition of ubiquitin as a factor in disease progression, either as part of the virus neuroinvasive program or as a cellular defense mechanism that is overcome by the DUB. Although several substrates of the DUB have been identified, how this enzyme promotes the potent neuroinvasive property of these viruses is unknown (23–30). To elucidate the role of the DUB, we set out to examine the role of ubiquitination during neuroinvasive herpesvirus infection. We report that ubiquitin addition and removal from a conserved lysine residue in pUL36 switches PRV between two invasive states that act successively to promote virus dissemination into the peripheral nervous system. Supporting evidence that the “ubiquitin switch” is conserved in HSV-1, and possibly all neuroinvasive alphaherpesviruses, is provided.
Results
Ubiquitination Promotes Retrograde Delivery of PRV Capsids to Cell Bodies of Sensory Neurons.
The requirement of the pUL36 DUB during neuroinvasion may be either a protective mechanism that stabilizes substrates by maintaining them in an unmodified state, or part of a regulatory mechanism that switches proteins between ubiquitinated and nonubiquitinated states. To discriminate between these possibilities, ubiquitination was inhibited during PRV infection in a neuron culture model (Fig. 1A). Upon infection of primary dorsal root ganglion (DRG) sensory neurons that were pretreated with PYR41, an inhibitor of the E1 ubiquitin activating enzyme, the delivery of PRV capsids to the nuclear rim was significantly reduced compared with the vehicle-treated control (Fig. 1B) (31). In contrast, interference was not observed in an epithelial cell line nor following treatment of neurons with an inhibitor of the related E1 activating enzyme of the ubiquitin-like modifier, NEDD8 (Fig. 1 B and C) (32). These findings indicated a specific role for ubiquitination in neurons during infection. Given that viral protein pUL36 is an effector of the retrograde axonal transport that delivers capsids to the neural soma, we investigated whether pUL36 was a target of ubiquitination during infection (2). A previous report indicated that pUL36 is ubiquitinated during transient overexpression when the DUB is inactivated by a catalytic mutation (23); this also proved true during infection of epithelial cells (Fig. 1D). In contrast, there was no evidence of pUL36 neddylation in this context, despite the prior finding that the pUL36 DUB can cleave NEDD8 from substrates to promote viral DNA replication (26).
Fig. 1.

Ubiquitination promotes capsid delivery to neuronal nuclei. (A) Diagram of capsid retrograde axonal transport and nuclear rim delivery. (B) Representative images of capsids (UL25/mCherry tag) at nuclear rims of cultured DRG (Top) with estimates of capsid numbers at nuclear rims at 3–4 hpi (Bottom) following pretreatment with DMSO (vehicle), PYR41 (10 µM), or MLN4924 (1 µM). Mean ± SD are shown (***P < 0.001 based on Tukey’s multiple-comparison test). (Scale bar: 10 μm.) (C) Estimates of capsid numbers at nuclear rims at 3–4 hpi following infection of PK15 epithelial cells. (D) pUL36 was immunoprecipitated from PK15 cells either mock-infected or infected with PRV expressing a GFP-pUL36 fusion at 8 hpi and probed for GFP-pUL36, ubiquitin (Ub), or NEDD8. Input lysates were probed for GFP-pUL36 and a cellular dynactin component (p150) as loading control.
A Conserved Lysine in pUL36 Is a Critical Virulence Determinant.
Mutational analysis was used to initially examine whether pUL36 ubiquitination promotes infection. Four lysines, the site of ubiquitin addition, that are invariant across the pUL36 homologs of seven neuroinvasive herpesviruses were individually mutated to arginine to prevent potential ubiquitination while maintaining the positive charge of the amino acid side chain (Fig. 2A). The four recombinant PRV strains propagated efficiently in epithelial cells, but two (K442R and K1613R) exhibited moderate reductions in spread (Fig. 2B). In contrast, defects associated with the K442R and K1613R mutations were profound during infection of mice (Fig. 2C). K442R-infected animals did not present symptoms during the course of the experiment, which was terminated at 336 h postinfection (hpi). The K1613R mutant was severely attenuated relative to the wild type (WT), K963R, and K1299R viruses. Genetic repair of the K442R and K1613R mutations restored virus spread and virulence to WT levels (Fig. 2 B and C).
Fig. 2.

A conserved lysine in pUL36 is a virulence determinant that sustains retrograde axonal transport. (A) Illustration of PRV pUL36. Positions of lysine residues are noted with vertical bars, and the DUB is shown in light gray with the position of the active-site cysteine indicated. Four lysines conserved across seven neuroinvasive herpesviruses are shown with local sequence alignments (GenBank JF797219, AJ004801, AY665713, NC_001348, NC_004812, GU734771, and Z86099). (B) Spread of viruses in PK15 epithelial cells (n = 150 per sample; mean ± SD shown in red; ***P < 0.001 based on Tukey’s multiple-comparison test; rep, genetic repair). (C) Kaplan–Meier presentation of mouse survival following intranasal instillation of 106 plaque-forming units (PFU) of PRV (n = 10 for K442R and K1613R; n = 5 for all others). (D) Frequency of aberrant (nonretrograde) PRV axonal transport in cultured DRG sensory neurons 30–60 min postinfection (three independent experiments; n > 100 viral particles per sample; mean values ± SEM; *P < 0.05, two-tailed unpaired t test). (E) The number of fluorescent capsids at nuclear rims of cultured DRG (Left, representative images) was estimated (Right) at 3–4 hpi. (Scale bar: 10 μm.)
To examine whether the virulence defect of the K442R mutant correlated with a reduced capacity to infect neurons, capsid retrograde axonal transport in DRG cultured ex vivo was monitored using live-cell fluorescence microscopy (Fig. 2D). In contrast to WT, K1613R, or genetically repaired K442 virus, capsids of the K442R mutant displayed aberrant motion in the form of frequent stops and misdirected (anterograde) transport that together impaired the long-distance delivery of capsids to the cell body (Fig. 2D and Movie S1). No corresponding defect was observed during epithelial infection (Fig. S1). The reduction in K442R capsid targeting to the neural soma phenocopied broad inhibition of ubiquitination during WT infection, implicating ubiquitin addition to pUL36 residue K442 as instrumental to neuronal infection (Figs. 1B and 2E).
Fig. S1.

K442 is dispensable for nuclear rim delivery and propagation in epithelial cells. (A) The number of fluorescent capsids at nuclear rims was estimated 2 hpi in PK15 cells. Representative images document that while the majority of input viral particles remain in proximity of cell surfaces, a fraction have traversed the cytoplasm and are now localized at the nuclear periphery. Mean ± SD of capsid fluorescence at nuclear rims are shown with statistics based on an unpaired t test. (Scale bar: 10 μm.) (B) Propagation kinetics of the K442R mutant on PK15 cells (MOI 10) relative to WT and repaired PRV. PFU, plaque-forming units.
The HSV-1 and PRV pUL36 DUBs Regulate the Ubiquitin State of Several Viral Proteins, Including pUL36.
Mass spectrometry was used to determine whether PRV pUL36 K442 was, in fact, ubiquitinated during infection. The analysis measured the amount of ubiquitin-like proteins (either ubiquitin, NEDD8, or ISG15) present on virus proteins isolated from HeLa cells infected for 8 h with either HSV-1 or PRV, each encoding either a wild-type DUB (WT) or a DUB catalytic mutant (∆DUB) (Fig. S2). Modifications were detected as posttryptic di-glycine conjugates on lysine side chains (33). A ratio of ∆DUB:WT modification was determined for each detected virus tryptic peptide, with values greater than 1 indicating the peptide contained a lysine that was modified more frequently in the absence of DUB activity during infection (Dataset S1). A summary score of combined peptide ratios for individual virus proteins was calculated, and a Pearson’s correlation analysis indicated a strong positive relationship between the two viruses (r = 0.87; Fig. 3A). The protein with the highest ∆DUB:WT value was pUL36 itself, which had three targeted lysines in PRV: K442, K963, and K1453 (Fig. 3B). Although the modifications identified by mass spectrometry could be from either ubiquitin, NEDD8, or ISG15, posttranslational modification at these pUL36 sites was consistent with ubiquitin given that the DUB is not active against ISG15 (6, 26), and NEDD8 was not detected as a pUL36 conjugate during infection with the ∆DUB virus (Fig. 1D). K963 (K1294 in HSV-1) was not further examined as it was dispensable during PRV infection (Fig. 2 B and C) and was not enriched in the HSV-1 sample (Fig. 3C). K1453 (K1789 in HSV-1) was also not pursued, as although ubiquitination at this site was enriched in the ∆DUB sample during HSV-1 infection (Fig. 3C), the lysine is not conserved in other neuroinvasive herpesviruses including HSV-2, which notably encodes arginine at the equivalent position (Fig. S3). Only K442 (K636 in HSV-1) is positionally conserved across the neuroinvasive alphaherpesviruses, was ubiquitinated during both HSV-1 and PRV infection, and was hyperubiquitinated in HSV-1 and PRV in the absence of an active pUL36 DUB (Fig. 3 and Fig. S3). These results are consistent with the noted requirement for an active ubiquitination pathway during neuronal infection, with the critical substrate mapping to K442 of PRV pUL36.
Fig. S2.

Monitoring of infection by major capsid protein (VP5) expression. HSV-1– and PRV-infected HeLa lysates used for Fig. 3 were blotted for HSV-1 and PRV VP5 and α-tubulin (loading control). WT viruses (DUB+); DUB inactive viruses (DUB−; HSV-1 C40A and PRV C26A).
Fig. 3.

Ranking of ubiquitinated viral proteins as pUL36 DUB substrates. (A) Comparison of PRV and HSV-1 mass spectrometry datasets as detailed for PRV (B) and HSV-1 (C). Tryptic peptides containing lysine di-glycine conjugates from lysates of HeLa cells infected with WT or DUB-inactivated PRV or HSV-1 were immunoprecipitated and examined by mass spectrometry. For each WT and ΔDUB pair, a peptide ratio (ΔDUB/WT) was determined and a value of 1 was subtracted. A summary score for each viral protein was then calculated by summing its peptide ratios. Each point on the graph represents one viral protein summary score (A), with greater values indicating the tendency of the protein to be modified in the absence of DUB activity. Only proteins that had modified lysines in both PRV and HSV-1 were plotted. A Pearson correlation analysis indicated a positive relationship between the two viruses (r = 0.87). (B and C) Positive peptide ratios were summed for each corresponding protein and plotted in rank order from poorest DUB substrate (Top) to greatest (Bottom). Black bars indicate conserved substrates in PRV and HSV-1. *Proteins with summed ratios < 1 (i.e., modified but not DUB substrates). The number of di-glycine modified lysines detected per protein is indicated in parentheses. Insets show pUL36 peptides having a ratio > 1 (dotted vertical lines, significance threshold of 1.5).
Fig. S3.
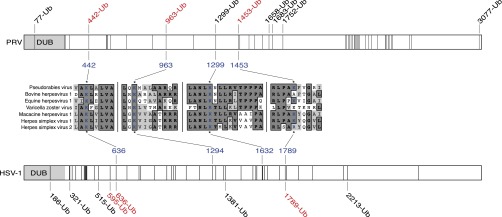
Local alignments of pUL36 lysines from seven neuroinvasive herpesviruses annotated with results from UbiScan mass spectrometry analysis of PRV and HSV-1 pUL36. Illustration of pUL36 lysine residues in PRV (Top, 49 lysines) and HSV-1 (Bottom, 48 lysines). Lysines are represented as vertical lines, with ubiquitinated lysines (Ub) indicated by amino acid position. Lysines that are DUB autocatalytic substrate sites are indicated in red. Local sequence alignments for the three lysines studied (PRV K442, K963, and K1299) are shown with corresponding HSV-1 lysines indicated. An alignment for a fourth lysine that is a DUB substrate in PRV and HSV-1, PRV K1453, is also provided. Alignment details: conserved amino acids are in bold with dark gray background, and amino acids with similar properties have a light gray background.
Lysine 442 Is Necessary for Retrograde Invasion and Spread Within the Nervous System.
To determine whether the defects associated with the K442R mutation translated to reduced neuroinvasion in a mammalian host, variants of PRV encoding an mCherry-capsid fusion that serves as a fluorescent reporter were injected intraocularly into the aqueous humor of the rat eye anterior chamber (AC) (Fig. 4A). Although not a natural route of infection, the AC model confines the initial infection to intraocular tissues that are bathed in the aqueous humor, including the iris, progressing to a well-defined invasion of the peripheral nervous system (PNS) by retrograde axonal transport (34). This model was previously used to document the neuroinvasive defect of PRV encoding a catalytically inactive DUB (8). At 44 hpi, both the ∆DUB and K442R viruses had spread within the iris, approaching but not equivalent to WT infection levels (Fig. 4B). Neuroinvasion was monitored by imaging the innervating ipsilateral superior cervical ganglion (SCG) at 48 hpi, which was sufficient time for WT virus to infect this autonomic ganglion of the PNS (Fig. 4C). Spread to SCG neurons only occurred following PRV replication in innervated tissues of the eye, as a defective PRV mutant (∆UL25) that cannot spread cell to cell produced negligible infection of the SCG, indicating that axon terminals in the eye are not exposed to the aqueous humor (Fig. 4C) (35). The ∆DUB virus was incapable of transmission to the SCG despite productive replication in the iris, which is consistent with the prior observation that DUB activity is required for PRV spread from the AC to the brain, and demonstrates that this established CNS neuroinvasive defect is due to an upstream loss of PNS neuroinvasion (7, 8). The K442R virus phenocopied the DUB mutant demonstrating that ubiquitination, in addition to deubiquitination, at K442 is critical for invasion of the PNS. Intraocular AC injection of repaired viruses resulted in infection of the SCG that was indistinguishable from the WT (Fig. 4C).
Fig. 4.

The herpes DUB and lysine substrate promote serial stages of neuroinvasion. (A and D) Two models of PRV retrograde axonal transport examined in this study (AC, anterior chamber; RGC, retinal ganglion cells; SC, superior colliculus; SCG, superior cervical ganglion). (A) Nerve endings are shielded by ocular tissue (iris) and not directly exposed to viral inoculum. (B and C) Representative images of the iris (B) and SCG (C) following intraocular injection of PRV strains encoding a mCherry-capsid reporter into the AC and retrograde transport to the SCG, with amount of infection based on fluorescent cell number indicated below. (D) Neurons and axons are directly exposed to viral inoculum. (E) Representative images and quantitation following intracranial injection into the SC and retrograde transmission to RGC of the contralateral eye. Microelectrode tracks are outlined in SC injection images to distinguish inoculum from viral spread. (Scale bars: 50 µm.)
To determine whether the loss of neuroinvasion with the K442R mutant was due to a defect in epithelial-neuron transmission, virus inoculum was injected directly into the superior colliculus (SC) of the brain, which receives axonal projections from retinal ganglion cells (RGCs) in the eye (Fig. 4D). In the absence of an epithelial barrier, the ∆DUB virus replicated and spread within the SC, reinforcing that DUB activity is dispensable for neurotropism but is critical for neuroinvasion (Fig. 4E) (8). Furthermore, the DUB mutant spread from the SC to the retina, confirming that the virus was competent to travel retrogradely in axons. The K442R mutant also remained neurotropic and spread within the SC, but did not transmit retrogradely within the axons of the optic nerve to the retina (Fig. 4E). The inability of the K442R mutant to transmit retrogradely from AC to SCG (indirect neuronal exposure) and from CNS to retina (direct neuronal exposure) was consistent with the noted defect in retrograde axonal transport (Fig. 2 D and E) and distinguished this neuroinvasive effector function from that of the DUB.
Discussion
Neurotropic herpesviruses such as HSV-1 and PRV exhibit profound, yet subtle, neuroinvasive properties. Infections typically begin in exposed mucosal tissue, with newly replicated viral particles rapidly transmitting within axons of innervating neurons and resulting in the establishment of lifelong latent infections in sensory and autonomic ganglia of the PNS. This feat is remarkable given that most unrelated viral infections occur one cell junction away from peripheral nerves, yet few viruses invade the nervous system. Of those that do, neuroinvasion is infrequent (i.e., poliovirus, measles virus, mumps virus, West Nile virus, Japanese encephalitic virus, and Chandipura virus) or occurs following an animal bite (i.e., rabies virus). In the case of the latter, mechanical tissue disruption at the site of inoculation bypasses intrinsic protective barriers in the skin and increases retrograde transport in damaged axons as part of a peripheral nerve repair response (36, 37). The concept that tissue trauma can increase the susceptibility of the nervous system to neurotropic viruses is best documented for poliovirus, which is rarely neuroinvasive except when provoked by tissue injury (38–40). The near impenetrability of the nervous system in healthy tissue indicates that effective intrinsic barriers to infection exist between peripheral tissues and the PNS. However, these barriers are seemingly ineffective against the neuroinvasive herpesviruses.
We report that PRV repurposes the cellular ubiquitin pathway as a means to switch the virus between two invasive states. Each state is capable of overcoming an intrinsic barrier that normally protects the PNS from infection, but neither state alone is sufficient to promote neuroinvasion (Fig. 5). A combination of genetic analysis and mass spectrometry data pinpointed K442 in the capsid-bound tegument protein, pUL36, as the relevant site of ubiquitination and deubiquitination that modulates the virus state. PRV, which is highly neurovirulent in rodent infection models, became avirulent and incapable of PNS invasion when encoding a single K442R codon change that prevented ubiquitination at this site. By capitalizing on a well-defined PRV ocular infection model, the opposing phenotypes of the ubiquitin switch became apparent: addition and removal of ubiquitin at K442 was consistent with alternating the virus between an axon-targeting mode within peripheral tissues (DUB dependent) and a sustained retrograde microtubule trafficking state following entry into axons (ubiquitination/K442 dependent).
Fig. 5.
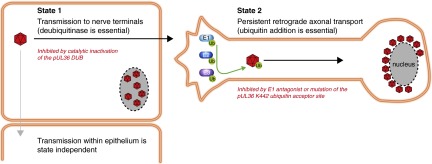
Ubiquitin switch model of herpesvirus neuroinvasion. The large tegument protein (pUL36) encodes a deubiquitinase (DUB) activity and a ubiquitin addition site (K442 in PRV), both of which are essential for virus invasion of the nervous system from peripheral tissues. The DUB and K442 acceptor site together constitute a molecular switch that alternates the virus between an axon targeting mode (state 1), and a sustained retrograde transport mode (state 2) that is required for the long-distance delivery of viral particles within nerve fibers to the peripheral ganglia.
The absolute requirement of the ubiquitin switch in coordinating the larger neuroinvasive phenotype indicates that the intrinsic barriers protecting nerve endings in peripheral tissues are diametrically opposed, and we suggest that this opposition hinders the evolution of neuroinvasive properties, as virus adaptations that overcome one barrier will be detrimental to the ability of a virus to overcome the opposing barrier. Given the role of pUL36 in microtubule-based transport, it seems likely that microtubule trafficking may be the source of the barrier opposition (2). Preventing ubiquitination at K442 moderately reduced viral spread in nonneuronal cells based on plaque assay, but severely impaired microtubule-based retrograde delivery of PRV in axons in explanted sensory ganglia and eliminated retrograde spread in vivo. The enhancement of retrograde microtubule transport that benefits PRV axonal transport is likely detrimental for spread from epithelia to nerve endings, as indicated by the ∆DUB phenotype, possibly by misdirecting virions from sites of epithelial/axonal contact. The benefit afforded by the DUB in epithelial cells, why the DUB activity is not detrimental within axons, and identifying the E3 ubiquitin ligase that targets pUL36 are important topics for future investigation.
Because the DUB and K442 are conserved elements of pUL36 across the neuroinvasive herpesviruses, the robust neuroinvasion exhibited by these Alphaherpesvirinae subfamily members are likely based upon the ubiquitin switch described here with PRV. In particular, the comparative mass spectrometry analysis of HSV-1 and PRV ubiquitination included in this report indicates that the conserved K442 site (K636 in HSV-1) is ubiquitinated and serves as a DUB substrate for both viruses. Thus, the ubiquitin switch provides a framework for understanding how a subset of herpesviruses are capable of routinely invading the mammalian nervous system.
Materials and Methods
Cells and Virus Strains.
Vero and PK15 epithelial cell lines were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% (vol/vol) bovine growth serum. HeLa cells were freshly obtained from the ATCC and maintained in DMEM supplemented with 10% (vol/vol) FBS. Bacterial artificial chromosome (BAC) infectious clones derived from PRV pBecker3 were transfected into PK15 cells as described previously (41). HSV-1 infectious clones derived from the pYEbac102 BAC (42) were transfected into Vero-Cre cells as previously described (4). The resultant virus stocks were passaged once to create working stocks. Titers and single-step propagation kinetics were determined by plaque assays on PK15 (PRV) or Vero (HSV-1) cells as described previously (4, 43) (Tables S1 and S2).
Table S1.
Strains used in this study
| Strain | mCherry* | GFP | Encoded UL36 product† | Titer, PFU/mL | Source |
| PRV-GS999 | — | — | WT | 3.0 × 109 | Smith and Enquist (41) |
| PRV-GS4957 | — | — | K442R | 1.0 × 109 | Current study |
| PRV-GS5951 | — | — | K442 (GS4957 repair) | 3.0 × 109 | Current study |
| PRV-GS5317 | — | — | K1613R | 6.3 × 108 | Current study |
| PRV-GS5531 | — | — | K1613 (GS5317 repair) | 1.6 × 109 | Current study |
| PRV-GS5380 | — | — | K963 | 2.7 × 109 | Current study |
| PRV-GS5381 | — | — | K1299 | 1.4 × 109 | Current study |
| PRV-GS4284 | pUL25 | — | WT | 2.2 × 109 | Bohannon et al. (43) |
| PRV-GS4747 | pUL25 | — | C26A | 2.1 × 108 | Current study |
| PRV-GS5971 | pUL25 | — | C26 (GS4747 repair) | 2.5 × 109 | Current study |
| PRV-GS5112 | pUL25 | — | K442R | 1.1 × 109 | Current study |
| PRV-GS5933 | pUL25 | — | K442 (GS5112 repair) | 2.7 × 109 | Current study |
| PRV-GS5270 | pUL25 | — | K1613R | 3.0 × 108 | Current study |
| PRV-GS5507 | pUL25 | GFP-pUL36 | WT | 1.5 × 109 | Current study |
| PRV-GS5508 | pUL25 | GFP-pUL36 | C26A (∆DUB) | 3.5 × 108 | Current study |
| PRV-GS5555 | — | CMV > GFP‡ | WT | 1.4 × 108 | Current study |
| HSV1-GS4553 | pUL25 | — | WT | 4.8 × 108 | Current study |
| HSV1-GS5553 | pUL25 | — | C40A (∆DUB)§ | 4.5 × 108 | Current study |
mCherry was fused in-frame between pUL25 amino acids 42–43 for PRV and 50–51 for HSV-1.
Amino acid numbers are based on PRV strain Becker (GenBank JF797219) and HSV-1 strain F (GenBank GU734771 and KM222724).
Soluble GFP cassette driven by a CMV promoter was inserted into the US4 gene. This virus is also deleted for the UL25 gene.
The catalytic cysteine at position 40 has been previously referred to as C65 based on an upstream in-frame ATG being assigned amino acid position 1.
Table S2.
En passant primer pairs used to construct PRV and HSV-1 recombinant viruses made for this study
| Strain | PCR template | Primers |
| GS4957, GS5112 | pEP-KanS2 | 5′-GCGCTGCTGGACGTGGCCGGCGACGACGCGTCCGCGCCCGTGGCCAGGCTGGCGCTGGTCGCGAGGATGACGACGATAAGTAGGG |
| 5′-GGCCTCGTTCGCGGGCCGCGCGACGCGCGCCGCGACCAGCGCCAGCCTGGCCACGGGCGCGGACAACCAATTAACCAATTCTGATTAG | ||
| GS5951, GS5933 | pEP-KanS2 | 5′-GCGCTGCTGGACGTGGCCGGCGACGACGCGTCCGCGCCCGTGGCCAAGCTGGCGCTGGTCGCGAGGATGACGACGATAAGTAGGG |
| 5′-GGCCTCGTTCGCGGGCCGCGCGACGCGCGCCGCGACCAGCGCCAGCTTGGCCACGGGCGCGGACAACCAATTAACCAATTCTGATTAG | ||
| GS5317, GS5270 | pEP-KanS2 | 5′-AACGTGCCCGTGGAGTACCAGGTGTGCTACCGCGCGGTCGGCGACAGACTGGCGGCCATGCTGAGGATGACGACGATAAGTAGGG |
| 5′-CGGGCGCAGCTCGGCGCCCTCGTCGGTGCACAGCATGGCCGCCAGTCTGTCGCCGACCGCGCGCAACCAATTAACCAATTCTGATTAG | ||
| GS5531 | pEP-KanS2 | 5′-AACGTGCCCGTGGAGTACCAGGTGTGCTACCGCGCGGTCGGCGACAAGCTGGCGGCCATGCTGAGGATGACGACGATAAGTAGGG |
| 5′-CGGGCGCAGCTCGGCGCCCTCGTCGGTGCACAGCATGGCCGCCAGCTTGTCGCCGACCGCGCGCAACCAATTAACCAATTCTGATTAG | ||
| GS5380 | pEP-KanS2 | 5′-CTGGAGGTGGCGCGCGCGGCCGCCGTGGAGGAGCCGCAGCTGCAGAGAATGGCGCTCGCGGCGAGGATGACGACGATAAGTAGGG |
| 5′-GGCCGCCTCCAGCTCGCCGACGCGCTGGCGCGCCGCGAGCGCCATTCTCTGCAGCTGCGGCTCCAACCAATTAACCAATTCTGATTAG | ||
| GS5381 | pEP-KanS2 | 5′-GCGGCGGCGCGCGACCGCGACGGGGCCCTCGAGCTGGCCAACCTCAGAAACCTGCTGCGGGTGAGGATGACGACGATAAGTAGGG |
| 5′-GTCGATGTTGAGCGAGGCCGGCGGCGGCGTCACCCGCAGCAGGTTTCTGAGGTTGGCCAGCTCCAACCAATTAACCAATTCTGATTAG | ||
| GS4747, GS5508 | pEP-KanS2 | 5′-GGCTATCGTAATCAGTATGACCCCGACCTGGGGCCCGGGTCGGGCGTCTCAGCGCTGCGCTCCTCCCTCAGGATGACGACGATAAGTAGGG |
| 5′-CTCCACGCCGTTCGTGAAGACCAGGCGCAGGAAGGAGAGGGAGGAGCGCAGAGCTGAGACGCCCGACCCCAACCAATTAACCAATTCTGATTAG | ||
| GS5971 | pEP-KanS2 | 5′-TCAGTATGACCCCGACCTGGGGCCCGGGTCGGGCGTCTCGTGCCTGCGCTCCTCCCTCTCCTTAGGATGACGACGATAAGTAGGG |
| 5′-TCGTGAAGACCAGGCGCAGGAAGGAGAGGGAGGAGCGCAGGCACGAGACGCCCGACCCGGGCCCAACCAATTAACCAATTCTGATTAG | ||
| GS5507, GS5508 | pEP-EGFP-in | 5′-AAAGATTTTTCCCCCACGCGCGTGTGTTATTTCAGCCATGGTGAGCAAGGGCGAGGAG |
| 5′-CATACTGATTACGATAGCCGACGACCACCGCGTCGGCCGTCTTGTACAGCTCGTCCATGC | ||
| GS5555 | pEP-KanS2 | 5′-GGTGGGCGCGGTGATAAGGCGGCGCGCGGCGGCGCGCGGCCCATGTAACAGTTTGGCGTCTCCAGGATGACGACGATAAGTAGGG |
| 5′-GCAGAGAAAGTACAGGAGGTCGTAGTCGCTGGAGACGCCAAACTGTTACATGGGCCGCGCGCCCAACCAATTAACCAATTCTGATTAG | ||
| GS4553, GS5553 | pEP-mCherry-in | 5′-GTCGCCCGTCTTTAACCTCCCCCGGGAGACGGCGGCGGAGGTGAGCAAGGGCGAGGAG |
| 5′-CGGCAGCCGCTGTGCGCTGGGCCTGTAGGACGACCACCTGCTTGTACAGCTCGTCCATGC | ||
| GS5553 | pEP-KanS2 | 5′-GGTGCTCGGAACCAGTTCGCGCCCGACCTGGAGCCGGGGGGGTCGGTATCGGCTATGCGCTCGTCGCTGTCCTTAGGATGACGACGATAAGTAGGG |
| 5′-GCGAGGGCCCACATCAAATATGAGGCTGAGAAAGGACAGCGACGAGCGCATAGCCGATACCGACCCCCCCGGCTCAACCAATTAACCAATTCTGATTAG |
Underlined sequence indicates PCR template homology.
Immunoprecipitation and Western Blotting.
PK15 cells were mock infected or infected at a multiplicity of infection (MOI) of 50 with recombinant viruses encoding GFP-pUL36 fusions. Lysates were prepared at 8 hpi as described previously (44). More details can be found in SI Materials and Methods.
Live-Cell Fluorescence Microscopy.
Imaging of axonal transport and capsid accumulation at neuronal nuclei was performed using primary cultured neurons from embryonic chicken [embryonic day 8 (E8) to E10] DRGs, as previously described (13). See SI Materials and Methods for further details.
In Vivo Inoculation of Virus.
All procedures conformed to NIH guidelines for work with laboratory animals and were approved by the Institutional Animal Care and Use Committee of the University of Nebraska, Lincoln. See SI Materials and Methods for further details.
Mass Spectrometry (UbiScan).
Approximately 2.0 × 108 HeLa cells were infected at MOI of 50 with either WT or DUB mutant PRV or HSV-1 and collected at 8 hpi for label-free UbiScan analysis (33). Detailed methods are in SI Materials and Methods. Raw and fold-change sorted data are combined in Excel format in Dataset S1.
SI Materials and Methods
Cells and Virus Strains.
Vero and PK15 epithelial cell lines were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% (vol/vol) bovine growth serum. HeLa cells were freshly obtained from the ATCC and maintained in DMEM supplemented with 10% (vol/vol) FBS. Bacterial artificial chromosome (BAC) infectious clones derived from PRV pBecker3 were transfected into PK15 cells as described previously (41). HSV-1 infectious clones derived from the pYEbac102 BAC (42) were transfected into Vero-Cre cells as previously described (4). The resultant virus stocks were passaged once to create working stocks, and titers were determined by plaque assays on PK15 (PRV) or Vero (HSV-1) cells as described previously (4, 43). The complete list of recombinant viruses used in this study is provided in Table S1, along with the primers used in their production by the en passant two-step recombination procedure in Table S2.
Viral Propagation Kinetics and Plaque Size Analysis.
Nonfluorescent PRV variants were used for single-step growth and plaque size analysis as previously described (43). Briefly, 4.3 × 105 PK15 cells were infected at a MOI of 10 and the supernatants and cell-associated virus were collected and frozen at −80 °C at 2, 5, 8, 12, and 24 h postinfection (hpi). Samples were thawed, sonicated, and titered on PK15 cells. For plaque diameter analysis, ∼200 PFU per virus were used to infect PK15 cells. At 72 hpi, plaquing medium was removed and cells were stained with neutral red dye (Sigma-Aldrich) and scanned at 2,000 dpi on a flatbed scanner. MetaMorph software (Molecular Devices) was used to measure plaque diameters. WT plaques were set to 100%, and mutant virus plaque diameters were plotted as %WT. Significance was determined using Tukey’s multiple-comparison test (***P < 0.001).
Immunoprecipitation and Western Blotting.
Approximately 4.3 × 105 PK15 cells were mock infected or infected at a MOI of 50 with recombinant PRV encoding GFP-pUL36 fusions (Table S1). Lysates were prepared as described previously (44). Briefly, cells were lysed 8 hpi in SDS lysis buffer [1% SDS, 0.5% Nonidet P-40, 10% (vol/vol) glycerol, 250 mM NaCl, 3 mM EDTA, 20 mM Tris (pH 8.0)] supplemented with 20 mM N-ethylmaleimide, 5 mM iodoacetate, 1 mM phenylmethylsulfonyl fluoride, 25 mM sodium fluoride, 1 mM sodium orthovanadate, and Sigma Protease Inhibitor Mixture (Sigma-Aldrich). Lysates were centrifuged at 4 °C for 10 min at 13,000 × g to remove cell debris. Supernatant was precleared using 10 μL of Protein G Plus Protein A Agarose Beads (Merck-Millipore). Immunoprecipitations were performed using the rabbit serum polyclonal anti-GFP (Life Technologies) conjugated to Protein G Plus Protein A Agarose Beads. Immunoprecipitated protein was eluted using 2× final sample buffer [62.5 mM Tris (pH 6.8), 10% (vol/vol) glycerol, 0.01% bromophenol blue, 2% (wt/vol) SDS, 10% (vol/vol) β-mercaptoethanol] and boiled for 5 min. Samples were separated on 5% SDS/PAGE. Proteins were transferred to 0.45-µm nitrocellulose membranes. Ubiquitin was detected using the mouse monoclonal anti-Ub clone FK2 (Enzo Life Sciences). NEDD8 was detected using rabbit serum polyclonal anti-NEDD8 (Cell Signaling Technology). GFP-pUL36 was detected using rabbit serum polyclonal anti-pUL36 raised against the PRV pUL36 peptide QPSQQRPPEAPWTWPEPRD (45). PTMScan lysates were separated on a 4–20% polyacrylamide gradient, and VP5 was detected using the mouse monoclonal anti-VP5 clone H1.4 for HSV-1 (Biodesign International) or mouse monoclonal anti-VP5 clone 3C10 for PRV (a generous gift from Lynn Enquist, Princeton University, Princeton). α-Tubulin was detected with the mouse monoclonal anti–α-tubulin clone DM1A (Abcam). Detection was performed using a LI-COR Odyssey Fc imaging system with donkey anti-rabbit IRDye 680RD or goat anti-mouse IRDye 800CW secondary antibodies (LI-COR Biosciences).
Live-Cell Fluorescence Microscopy.
Imaging of axonal transport and capsid accumulation at neuronal nuclei was performed using primary cultured neurons from embryonic chicken (E8–E10) dorsal root ganglia (DRGs), as previously described (13). Briefly, DRG explants were grown on poly-dl-ornithine and laminin-treated coverslips in 2 mL of F12 media (Invitrogen) containing nutrient mix: 0.08 g/mL BSA fraction V powder (VWR), 0.4 mg/mL crystalline bovine pancreas insulin (Sigma-Aldrich), 1:2,500 1.0 mg/mL sodium selenite (VWR), 4 μg/mL transferrin (Intercell Technology), and 5 μL of 0.05 mg/mL nerve growth factor (Sigma-Aldrich). Explants were cultured for 2–3 d before infection with PRV variants encoding the pUL25/mCherry fusion at 5.0 × 107 PFU (Table S1). Time-lapse images were captured using an inverted wide-field Nikon Eclipse TE2000-U microscope fitted with a 60×/1.4 numerical aperture (N.A.) objective and a CascadeII:512 electron-multiplying charge-coupled device (EM-CCD) (Photometrics). The microscope was housed in a 37 °C environmental box (In Vivo Scientific). Transport events were imaged from 30 to 60 min postinfection. Moving particles were monitored by time-lapse fluorescence microscopy at 10 frames per s (100-ms exposures) for 200 frames. Particle velocities were determined using the kymograph function of the MetaMorph software package (Molecular Devices). Kymographs were generated using the multiline tool with a width of 20 pixels and average background subtraction. Entire particle paths, whether moving, stalled, or reversing, were traced within the kymograph using the “Multi-line” tool. Data were filtered postanalysis to only include velocities of runs ≥0.2 µm. Fraction of time stopped, in anterograde motion, and in retrograde motion were calculated for each particle by dividing the total time the particle was either stopped or moving by the total time the particle was imaged. Greater than 100 particles were analyzed per virus for three replicate experiments, and an average value was calculated for each virus. Values reported represent the mean ± SEM of the average values obtained. Significance was determined using Student's two-tailed unpaired t test (*P < 0.05). Graphs were created in GraphPad Prism 4 (GraphPad Software).
For assessing delivery of capsids to the nucleus, either PK15 epithelial cells or DRG explants were infected with PRV variants encoding pUL25/mCherry (Table S1) and imaged between 2 and 3, and 3 and 4 hpi, respectively, using a Nikon Ti inverted microscope fitted with a 100×/1.45 N.A. objective (Nikon Instruments) and coupled to a CSU-W1 confocal head (Yokogawa Electric Corporation) and a CascadeII:1024 EM-CCD (Photometrics). Illumination was provided by a Sapphire 561 laser (Coherent) and custom laser launch (Solamere Technology Group, Inc.). Either differential interference contrast (DRG) or 1.0 μg/mL Hoechst 33342 (PK15) (Life Technologies) was used to identify nuclear periphery. MetaMorph software was used to estimate the number of fluorescent capsids per nuclear rim. First, a threshold was applied to remove background from the images, and next the total emissions from pUL25/mCherry capsids were measured using an elliptical region that included the nuclear periphery (nuclear interiors were not included as they were below threshold), and the integrated fluorescence intensity of each nuclear region was divided by the average intensity of three isolated intracellular capsids. The latter normalized the nuclear intensity values to a per-capsid equivalency. Ubiquitin E1 activating enzyme inhibitor (UBA1 inhibitor) PYR41 was purchased from Tocris, dissolved in DMSO, added to cells to a final concentration of 10 μM 1 h before infection, and maintained throughout the infection. NEDD8 E1 Activating Enzyme Inhibitor (NAE Inhibitor) MLN4924 was purchased from Boston Biochem, dissolved in DMSO, and added to cells to a final concentration of 1.0 μM 6 h before infection and maintained throughout the infection.
In Vivo Inoculation of Virus.
All procedures conformed to NIH guidelines for work with laboratory animals and were approved by the Institutional Animal Care and Use Committee of the University of Nebraska, Lincoln. Intranasal inoculation was performed as previously described (43). Briefly, male CD-1 mice (6 wk old; Charles River) were maintained for at least 2 wk under a 12:12 h light/dark cycle, two to three mice per cage with food and water freely available. Intranasal application of PRV was administered to animals anesthetized by 2.5–5.0% isoflurane inhalation. Viral stocks were maintained frozen at −80 °C and used immediately after being thawed. Each animal received 5 μL of PRV (0.5–1.0 × 106 PFU) in each nostril. Behavior was continuously video monitored with images captured every 10 min. Survival times postinoculation were rounded to the nearest hour. Intraocular and intracranial injections of male Long–Evans rats, isolation of nervous tissues, and processing for fluorescence imaging were as previously described (8).
Mass Spectrometry (UbiScan).
Approximately 2.0 × 108 HeLa cells were infected at MOI of 50 with either WT or DUB mutant PRV or HSV-1 (Table S1). At 8 hpi, cells were washed with cold PBS and lysed in urea lysis buffer [20 mM Hepes (pH 8.0), 9 M urea, 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, and 1 mM β-glycerol-phosphate]. Lysates were sonicated to shear genomic DNA and centrifuged at 4 °C for 15 min at 20,000 × g to pellet cell debris. The supernatants were frozen at −80 °C and shipped on dry ice overnight to Cell Signaling Technology for label-free UbiScan analysis (33). The cell extracts were digested with trypsin and lyophilized via solid-phase extraction on a C18 column. Reconstituted peptides were incubated with a K-ε-GG remnant motif antibody (Cell Signaling Technology) and immobilized on protein A-agarose beads. The resin was washed to remove unbound peptides, and peptides immunoprecipitated with the K-ε-GG remnant motif antibody were eluted. Each sample was divided in half used for independent liquid chromatography–tandem mass spectrometry (LC-MS/MS) runs. Peptides were loaded directly onto a 10 cm × 75-μm PicoFrit capillary column packed with Magic C18 AQ reversed-phase resin. The column was developed with a 72-min linear gradient of acetonitrile in 0.125% formic acid delivered at 280 nL/min. LC-MS/MS analysis was performed on a LTQ-Orbitrap-Elite, using electrospray ionization–collision-induced dissociation. MS parameter settings were as follows: MS run time, 96 min; MS1 scan range (300–1500); top 20 MS/MS (min signal 500, isolation width 2.0, normalized coll. energy 35, activation-Q 0.25, activation time 20, lock mass 371.101237, charge state rejection enabled, charge state 1+ rejected, dynamic exclusion enabled, repeat count 1, repeat duration 35, exclusion list size 500, exclusion duration 40, exclusion mass width relative to mass, exclusion mass width 10 ppm). MS/MS spectra were evaluated using SEQUEST 3G and the SORCERER 2 platform from Sage-N Research (version 4.0). Searches were performed against the human database (National Center for Biotechnology Information release date, 6/27/2011); Suid herpesvirus 1 strain Becker database (release date, 11/2/2011) for PRV samples; and human herpesvirus 1 strain F database (release date, 4/27/2010) for HSV-1 samples with a mass accuracy of ±50 ppm for precursor ions and 1 Da for product ions. Results were filtered with mass accuracy of ±5 ppm on precursor ions and presence of the intended modification K-ε-GG. A 5% default false-positive rate was used to filter the SORCERER results. Results were further filtered based on the analytical variation (percent coefficient of variation) and intensity values being above background in at least one sample. Raw and fold-change sorted data are combined in Dataset S1.
Supplementary Material
Acknowledgments
We thank Jonathan Leis and Gina Daniel for helpful discussions of the data and approach, Jenifer Klabis for assistance with recombinant virus production, Riley Junell and Anne Fischer for technical assistance, and Jeffrey Silva and Charles Farnsworth from Cell Signaling Technology for mass spectrometry. Sequencing services were performed at the Northwestern University Genomics Core Facility. This work was funded by NIH Grants R01 AI056346 (to G.A.S.) and R01 NS077003 (to G.E.P. and P.J.S.). N.J.H. received support from Viral Replication Training Grant T32AI060523 from the NIH.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1512559112/-/DCSupplemental.
References
- 1.Koyuncu OO, Song R, Greco TM, Cristea IM, Enquist LW. The number of alphaherpesvirus particles infecting axons and the axonal protein repertoire determines the outcome of neuronal infection. MBio. 2015;6(2):e00276-15. doi: 10.1128/mBio.00276-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zaichick SV, et al. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe. 2013;13(2):193–203. doi: 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koyuncu OO, Perlman DH, Enquist LW. Efficient retrograde transport of pseudorabies virus within neurons requires local protein synthesis in axons. Cell Host Microbe. 2013;13(1):54–66. doi: 10.1016/j.chom.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antinone SE, Smith GA. Retrograde axon transport of herpes simplex virus and pseudorabies virus: A live-cell comparative analysis. J Virol. 2010;84(3):1504–1512. doi: 10.1128/JVI.02029-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aggarwal A, et al. Ultrastructural visualization of individual tegument protein dissociation during entry of herpes simplex virus 1 into human and rat dorsal root ganglion neurons. J Virol. 2012;86(11):6123–6137. doi: 10.1128/JVI.07016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kattenhorn LM, Korbel GA, Kessler BM, Spooner E, Ploegh HL. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol Cell. 2005;19(4):547–557. doi: 10.1016/j.molcel.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Böttcher S, et al. Mutagenesis of the active-site cysteine in the ubiquitin-specific protease contained in large tegument protein pUL36 of pseudorabies virus impairs viral replication in vitro and neuroinvasion in vivo. J Virol. 2008;82(12):6009–6016. doi: 10.1128/JVI.00280-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JI, et al. A herpesvirus encoded deubiquitinase is a novel neuroinvasive determinant. PLoS Pathog. 2009;5(4):e1000387. doi: 10.1371/journal.ppat.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlieker C, Korbel GA, Kattenhorn LM, Ploegh HL. A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J Virol. 2005;79(24):15582–15585. doi: 10.1128/JVI.79.24.15582-15585.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coller KE, Lee JI, Ueda A, Smith GA. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J Virol. 2007;81(21):11790–11797. doi: 10.1128/JVI.01113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardone G, et al. The UL36 tegument protein of herpes simplex virus 1 has a composite binding site at the capsid vertices. J Virol. 2012;86(8):4058–4064. doi: 10.1128/JVI.00012-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan WH, et al. The large tegument protein pUL36 is essential for formation of the capsid vertex-specific component at the capsid-tegument interface of herpes simplex virus 1. J Virol. 2015;89(3):1502–1511. doi: 10.1128/JVI.02887-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luxton GW, et al. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc Natl Acad Sci USA. 2005;102(16):5832–5837. doi: 10.1073/pnas.0500803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Granzow H, Klupp BG, Mettenleiter TC. Entry of pseudorabies virus: An immunogold-labeling study. J Virol. 2005;79(5):3200–3205. doi: 10.1128/JVI.79.5.3200-3205.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knipe DM, Batterson W, Nosal C, Roizman B, Buchan A. Molecular genetics of herpes simplex virus. VI. Characterization of a temperature-sensitive mutant defective in the expression of all early viral gene products. J Virol. 1981;38(2):539–547. doi: 10.1128/jvi.38.2.539-547.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Batterson W, Furlong D, Roizman B. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J Virol. 1983;45(1):397–407. doi: 10.1128/jvi.45.1.397-407.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jovasevic V, Liang L, Roizman B. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J Virol. 2008;82(7):3311–3319. doi: 10.1128/JVI.01919-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abaitua F, Hollinshead M, Bolstad M, Crump CM, O’Hare P. A Nuclear localization signal in herpesvirus protein VP1-2 is essential for infection via capsid routing to the nuclear pore. J Virol. 2012;86(17):8998–9014. doi: 10.1128/JVI.01209-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schipke J, et al. The C-terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J Virol. 2012;86(7):3682–3700. doi: 10.1128/JVI.06432-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desai PJ. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J Virol. 2000;74(24):11608–11618. doi: 10.1128/jvi.74.24.11608-11618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuchs W, Klupp BG, Granzow H, Mettenleiter TC. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J Virol. 2004;78(21):11879–11889. doi: 10.1128/JVI.78.21.11879-11889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J Virol. 2006;80(1):201–209. doi: 10.1128/JVI.80.1.201-209.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolstad M, Abaitua F, Crump CM, O’Hare P. Autocatalytic activity of the ubiquitin-specific protease domain of herpes simplex virus 1 VP1-2. J Virol. 2011;85(17):8738–8751. doi: 10.1128/JVI.00798-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang S, Wang K, Li J, Zheng C. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J Virol. 2013;87(21):11851–11860. doi: 10.1128/JVI.01211-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saito S, et al. Epstein-Barr virus deubiquitinase down-regulates TRAF6-mediated NF-κB signaling during productive replication. J Virol. 2013;87(7):4060–4070. doi: 10.1128/JVI.02020-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gastaldello S, et al. A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-RING ligases. Nat Cell Biol. 2010;12(4):351–361. doi: 10.1038/ncb2035. [DOI] [PubMed] [Google Scholar]
- 27.Whitehurst CB, et al. The Epstein-Barr virus (EBV) deubiquitinating enzyme BPLF1 reduces EBV ribonucleotide reductase activity. J Virol. 2009;83(9):4345–4353. doi: 10.1128/JVI.02195-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inn KS, et al. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J Virol. 2011;85(20):10899–10904. doi: 10.1128/JVI.00690-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitehurst CB, Vaziri C, Shackelford J, Pagano JS. Epstein-Barr virus BPLF1 deubiquitinates PCNA and attenuates polymerase η recruitment to DNA damage sites. J Virol. 2012;86(15):8097–8106. doi: 10.1128/JVI.00588-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Gent M, et al. Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog. 2014;10(2):e1003960. doi: 10.1371/journal.ppat.1003960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y, et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007;67(19):9472–9481. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- 32.Soucy TA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 33.Kim W, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44(2):325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strack AM, Loewy AD. Pseudorabies virus: A highly specific transneuronal cell body marker in the sympathetic nervous system. J Neurosci. 1990;10(7):2139–2147. doi: 10.1523/JNEUROSCI.10-07-02139.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McNab AR, et al. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J Virol. 1998;72(2):1060–1070. doi: 10.1128/jvi.72.2.1060-1070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanz S, et al. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron. 2003;40(6):1095–1104. doi: 10.1016/s0896-6273(03)00770-0. [DOI] [PubMed] [Google Scholar]
- 37.Curtis R, et al. Retrograde axonal transport of ciliary neurotrophic factor is increased by peripheral nerve injury. Nature. 1993;365(6443):253–255. doi: 10.1038/365253a0. [DOI] [PubMed] [Google Scholar]
- 38.Lancaster KZ, Pfeiffer JK. Limited trafficking of a neurotropic virus through inefficient retrograde axonal transport and the type I interferon response. PLoS Pathog. 2010;6(3):e1000791. doi: 10.1371/journal.ppat.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gromeier M, Wimmer E. Mechanism of injury-provoked poliomyelitis. J Virol. 1998;72(6):5056–5060. doi: 10.1128/jvi.72.6.5056-5060.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strebel PM, Ion-Nedelcu N, Baughman AL, Sutter RW, Cochi SL. Intramuscular injections within 30 days of immunization with oral poliovirus vaccine—a risk factor for vaccine-associated paralytic poliomyelitis. N Engl J Med. 1995;332(8):500–506. doi: 10.1056/NEJM199502233320804. [DOI] [PubMed] [Google Scholar]
- 41.Smith GA, Enquist LW. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc Natl Acad Sci USA. 2000;97(9):4873–4878. doi: 10.1073/pnas.080502497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: Viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol. 2003;77(2):1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bohannon KP, Sollars PJ, Pickard GE, Smith GA. Fusion of a fluorescent protein to the pUL25 minor capsid protein of pseudorabies virus allows live-cell capsid imaging with negligible impact on infection. J Gen Virol. 2012;93(Pt 1):124–129. doi: 10.1099/vir.0.036145-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hwang J, Kalejta RF. In vivo analysis of protein sumoylation induced by a viral protein: Detection of HCMV pp71-induced Daxx sumoylation. Methods. 2011;55(2):160–165. doi: 10.1016/j.ymeth.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leelawong M, Lee JI, Smith GA. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J Virol. 2012;86(11):6303–6314. doi: 10.1128/JVI.07051-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.