

Abstract
With approximately 3% of the world’s population (170 million people) infected with the hepatitis C virus (HCV), the WHO has declared HCV a global health problem. Upon acute infection about 50%-80% of subjects develop chronic hepatitis with viral persistence being at risk to develop liver cirrhosis and hepatocellular carcinoma. One characteristic of HCV is its enormous sequence diversity, which represents a significant hurdle to the development of both effective vaccines as well as to novel therapeutic interventions. Due to a polymerase that lacks a proofreading function HCV presents with a high rate of evolution, which enables rapid adaptation to a new environment including an activated immune system upon acute infection. Similarly, novel drugs designed to specifically inhibit viral proteins will face the potential problem of rapid selection of drug resistance mutations. This review focuses on the sequence diversity of HCV, the driving forces of evolution and the impact on immune control and treatment response. An important feature of any therapeutic or prophylactic intervention will be an efficient attack of a structurally or functionally important region in the viral protein. The understanding of the driving forces, but also the limits of viral evolution, will be fundamental for the design of novel therapies.
Keywords: Hepatitis C Virus, Evolution, Escape, Drug resistance, Selection
INTRODUCTION
With approximately 3% of the world’s population (170 million people) infected with the hepatitis C virus (HCV), the WHO has declared HCV a global health problem[1]. Upon acute infection about 50%-80% of subjects develop chronic hepatitis with viral persistence being at risk to develop liver cirrhosis and hepatocellular carcinoma. The current standard treatment for chronic HCV infection is based on a combination of pegylated interferon alpha and ribavirin[2,3]. However, this combination is successful in only 50% of all subjects with response rates substantially influenced by the infecting viral genotype. Moreover, the side effects and costs of treatment further limit the use in many regions. Alternative treatment approaches including immunotherapeutic interventions are, therefore, highly warranted. One characteristic of HCV is its enormous sequence diversity, which represents a significant hurdle to the development of both effective vaccines as well as to novel therapeutic interventions. This review focuses on the sequence diversity of HCV, the driving forces of evolution and the impact on immune and control and treatment response.
GENOME STRUCTURE
The HCV genome is an RNA molecule of approximately 9600 nucleotides structured in a coding region that contains one large open reading frame and flanked by non-translated regions at the 5’ and 3’ ends. The polyprotein is cleaved into structural (core, envelope 1 and 2) and non-structural proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B) with one additional small protein at the junction between the structural and non-structural elements (p7 protein)[4]. Until the recent development of an in vitro replication system that produces infectious viral particles[5-7] many of the protein functions have been studied in sub-genomic replication systems or with purified protein after expression.
The 5’ NTR forms a highly structured RNA element that contains an internal ribosomal entry site (IRES) that allows interaction with the 40S ribosomal subunit and initiation of cap-independent translation of the viral RNA[8]. The polyprotein is translated as one large reading frame and subsequently cleaved by host-cellular proteases and virally encoded proteases into the individual proteins[9]. The structural proteins core, envelope 1 and envelope 2 are cleaved by host-cellular proteases. Processing of these proteins is believed to take place in a membrane associated complex at the endoplasmic reticulum by signal peptidases[10]. The core protein forms the viral capsid, binds to the viral RNA and interacts with envelope proteins to form viral particles. Different receptors have been suggested for the interaction of viral particles with the hepatocyte that mediates HCV entry including CD81, scavenger receptor class-B type-I (SR-BI), low-density lipoprotein receptor (LDL), mannose binding lectins (L-SIGN and DC-SIGN) and glycosaminoglycans[11]. E2 binds with high affinity to the extracellular loop of CD81, a tetraspanin that is expressed on different cell types including hepatocytes[12]. Cell entry of the virus is CD81 and SR-BI dependent, suggesting that these molecules serve as receptors or co-receptors for infection[13-16]. However, this interaction does not explain the hepatotropism as CD81 and SR-BI are not exclusively expressed on hepatocytes. Moreover, it has been shown that CD81 and SR-BI are necessary for cell entry of viral particles, but are not sufficient[16,17]. Some cell lines that express both proteins do not support viral entry. Recently a new candidate has been suggested that may close this gap. Claudin-1, a tight junction component that is highly expressed in the liver, was recently identified as a key factor in the late entry process[18].
These structural components of HCV are flanked by the non-structural proteins NS2 to NS5B. The function of one additional protein (p7) between these elements remains to be elucidated. It has been suggested that p7 forms an ion channel in planar lipid bilayers[19,20]. However, it is still unclear whether it is a virion component. NS2 contains an autoprotease, which cleaves the junction between NS2 and NS3[4]. NS3 is a multifunctional protein with a N-terminal protease domain and a C-terminal RNA helicase/NTPase domain. The NS3 protease cleaves the remaining non-structural proteins with NS4A as a cofactor for this activity. The NS3 RNA helicase/NTPase unwinds RNA and DNA[21]; however, its role during viral replication is unclear. The integral membrane protein NS4B is sufficient to induce membranous web formation and has been proposed to serve as a scaffold for replication complex assembly. The role of NS5A is again unclear. Numerous protein-protein interactions have been suggested including a role in silencing the host’s innate immune response and determining responsiveness to interferon alpha[22]. Some of these interactions are discussed later. NS5B encodes the viral RNA-dependent RNA polymerase. In the viral replication cycle the positive-strand RNA genome serves as a template to make a negative-strand intermediate, which then again serves as a template to produce multiple nascent genomes. Recently, one additional protein resulting from framshifted translation of the core protein has been identified (alternative reading frame protein ARFP)[23]. However, the function of this protein is unknown.
GENOTYPES
Phylogenetic analysis of HCV genomes revealed that sequences fall into different clusters. This observation led to a classification of HCV into different genotypes and a standardized nomenclature was proposed in a consensus paper in 1994[24]. The global distribution of HCV genotypes is regionally specific. The predominant genotype in most areas is genotype 1. However, some areas are almost exclusively infected with other genotypes. For example, the predominant genotype in Egypt is genotype 4 and the HCV epidemic in this country could be linked to parenteral treatment of schistosomiasis in the 1950s[25,26]. In some regions in Africa genotype 2 is more frequent[27]. In some areas of Asia, however, genotypes 3 and 6 are predominant[28-30]. Despite substantial sequence variation all genotypes share the same structure of linear genes of nearly identical size. The genotype specific variation of the different genes is remarkably consistent and has enabled many of the currently recognized variants of HCV to be provisionally classified based on partial sequences from subgenomic regions such as core/E1 and NS5B. The original nomenclature was recently updated further standardizing the nomenclature of existing variants[30]. Based on phylogenetic analysis a classification into 6 major genotypes was proposed and criteria for the designation of new HCV variants were formulated. These proposals provide an HCV nomenclature scheme for the three major public HCV sequence databases (Europe[31], USA[32] and Japan: http://s2as02.genes.nig.ac.jp/) and eliminate inconsistencies of the current classification procedures. HCV genotypes differ from each other by 31%-33% on the nucleotide level[33]. The genotypes are further divided into multiple epidemiologically distinct subtypes differing by 20%-25% from one other[33]. A phylogenetic tree depicting all published complete HCV genomes is presented in Figure 1. For many of the HCV subtypes, particularly for the less frequent ones, complete genome sequences are not available. The lack of sequence data for rare genotypes is profound prompting a sequencing initiative supported by the NIH to improve the HCV sequence databases[32].
Figure 1.
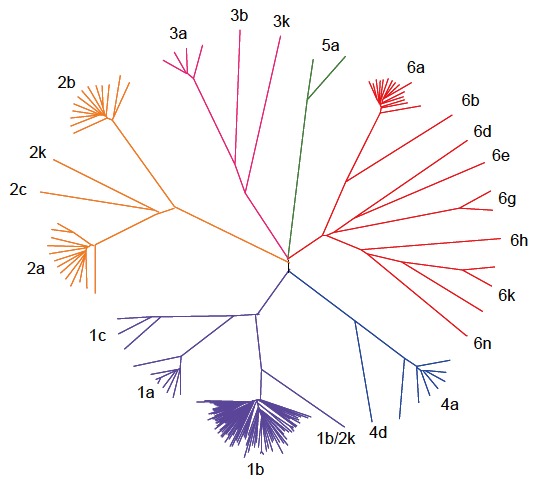
Published HCV Full Genomes. 169 full length HCV sequences available from the Los Alamos National Laboratory (LANL) HCV Sequence Database are illustrated in a phylogenetic tree. HCV sequences fall into six different clusters (genotype 1-6) and are further classified into subtypes. The sequence from one recombinant virus (genotype 1b/2k) is included.
SEQUENCE VARIATION
As a member of the flaviviridae, the virally encoded RNA polymerase of HCV lacks a proof reading function. Replication of this positive-stranded RNA genome is, therefore, characterized by ongoing error rates between 1 in 10000 and 1 in 100000 bp copied, which are typically found for RNA polymerases[34]. Together with a high turn-over rate of estimated 1012 virions per day[35], theoretically every possible mutation in every single position of the genome will be generated in one infected host every day. This high error rate is reflected in the generation of a heterogeneous, but closely related swarm of viruses within the same host referred to as quasispecies. The quasispecies nature of HCV can be best illustrated by sequence analysis of a short, but highly polymorphic region in envelope 2 designated as the hypervariable region 1 (HVR 1). Analysis of clonal sequences reveals that sequences of the viral population from the same subject are highly variable, but still phylogenetically closely related. In public databases normally the consensus sequence, as the most predominant residue at any given position within the quasispecies population, is presented. The quasispecies nature of HCV may have important consequences during a transmission event. Depending on the transmission route the number of transmitted viral RNA copies can be limited and may not represent the true complexity of the sequence diversity of the donor. This bottleneck phenomenon has been described for sexual transmission of HCV[36] and in the chimpanzee model[37]. However, the bottleneck could also be interpreted as selection of the optimal strain in the new host during the earliest infection events. Different aspects about the nature of the observed HCV sequence evolution have been published. The next section gives an overview of the different mechanisms.
DRIVING FORCES OF EVOLUTION
Genetic drift
Longitudinal analysis of isolates from subjects with chronic HCV infection calculated a mutation rate on the order of 1.5-2.0 × 10-3 nucleotide substitutions per site per genome per year[38,39]. In the model of neutral evolution mutations are selectively neutral[40]. The spread of these neutral mutations is mainly influenced by stochastic factors and is called genetic drift. As a consequence of this stochastic process even disadvantageous mutations can reach fixation when the virus circulates through a sufficiently small population. In turn, advantageous mutations are also affected by genetic drift when they are rare and are occasionally lost from the population. Several studies have described the rapid sequence drift of HCV over time[33]. The model of neutral evolution and the presumption that such diversification should occur at a constant rate over time provide a framework to estimate times of spread of HCV in specific transmission networks[41,42]. For example, a recent analysis of viral sequences obtained from an HCV and HIV outbreak in children at the Al-Fateh hospital in Libya, utilizing this molecular clock, demonstrated that the origin of the outbreak predated the year of 1998[43]. Noteworthy, this analysis excluded the possibility that the source virus was transmitted by foreign medical staff as suggested by local authorities.
Sequence drift has been suggested in a few studies as the major driving force of HCV evolution. An analysis by Allain et al suggested a dominant role in the evolution of envelope 2[44]. They analyzed clonal sequences of six different transmission pairs years after the transmission event. In this study the ratio of non-synonymous to synonymous mutations in the analyzed region did not support the hypothesis of positive or negative selection. The author’s, therefore, concluded that neutral evolution is a major component of the observed sequence diversity. Moreover, the authors did not find a correlation between the strength of the antibody response and the rate of evolution in these patients. However, in this study the strength of the humoral immune response was determined in serum collected years after the transmission event utilizing different HVR 1 peptides corresponding to autologous and heterologous sequences. Therefore, the true antibody response against the virus present during the acute phase of infection might have been underestimated. Another shortcoming of this study is that the inoculum sequence at the time of transmission was unknown and only a single time point years later was available making conclusions about the true evolutionary rate and kinetics difficult.
Positive and negative selection
In contrast to being neutral mutations may also be selected. Many mutations are probably disadvantageous or even deleterious for the virus and these variants are eliminated in a negative selection process. However, some mutations may not have an impact on replication capacity and a few may even be beneficial and confer a replication advantage. Variants harbouring these beneficial mutations will out compete for others in a dynamic process of continuous positive selection. Similar to other highly variable pathogens a complex process of continuous selection has been proposed for HCV[45]. Theoretically, infections with persistent viruses such as HIV and HCV have time to evolve within the same host before transmission to the next host and may adapt to the specific environment in an individual. The evolution of HCV may, therefore, be substantially influenced by host factors mediating selection pressure on the virus. Even though the consensus sequence may be close to the maximum of viral replication capacity at any one time, the existence of a large and diverse viral population allows rapid, adaptive changes in response to changes in the replication environment. Many variants that are beneficial in a new environment may already be present in a low frequency in the quasispecies population and subsequently out competes the existing dominant sequence. The impact of the quasispecies complexity on the clinical outcome can be profound. Farci et al analyzed sequences covering HVR1 obtained during the acute phase of infection from subjects who spontaneously resolved viremia and subjects who continued to chronic infection[46]. Spontaneous resolution of viremia was predicted by a decrease in quasispecies complexity during the first weeks of infection. In turn patients with viral persistence had increasing viral diversity suggesting a fast adaptation process to the new environment. A similar effect of quasispecies diversity on the outcome of treatment is discussed[47,48]. In recent years many studies have been published that aim to characterize the driving forces of this selection process.
Antibodies
The most variable region in the HCV genome is a short fragment spanning 27 amino acids of envelope 2 and is, therefore, designated the hypervariable region 1 (HVR 1). There is strong evidence that the profound sequence diversity in this region is the result of immune pressure by virus specific antibodies. Importantly, there is a close association between the observed sequence diversity in this region and the appearance of HCV specific antibodies in the sera of subjects with acute infection[49,50]. Patients suffering from common variable immunodeficiency (CVID) who present with hypogammaglobulinemia are not able to produce high titres of HCV specific antibodies and, therefore, are not able to mount humoral immune selection pressure. Analysis of sequences of HVR1 revealed that patients with hypogammaglobulinemia had significantly less amino acid substitutions in this region over time as compared to controls[51]. In a similar analysis, the rate of non-synonymous and synonymous mutations was compared between core and envelope in patients with and without CVID[52]. The rate of synonymous or silent mutations was similar in the core and envelope protein. In patients without CVID, as expected, the rate of non-synonymous mutations was much higher in envelope as compared to core, a protein that is known to be highly conserved. However, this high rate of non-synonymous mutations in envelope was not observed in patients with CVID suggesting that evolution is triggered by the presence of anti-HCV antibodies. In the chimpanzee model, it was demonstrated that a high turn-over rate is not sufficient to explain HVR1 sequence diversity. Only minor sequence variation was observed in this region upon serial infection with passage of an infectious HCV clone between 8 different animals[53]. Noteworthy, samples for the subsequent infection of the next animal were taken during the acute phase before antibodies became detectable. Again, this study indicates that this region in the envelope remains stable in the absence of antibodies despite high level viremia that was present in all animals during the acute phase of infection. Taken together, all these studies suggest that without immune selection pressure only minor sequence changes occur in HVR1.
The lack of an in vitro culture system has hampered direct evaluation of these putative escape mechanisms in the envelope protein. Recently, more elegant tools for this type of analysis became available by pseudotyping retroviral particles with HCV glycoproteins (HCVpp)[54-56]. Utilizing this technique, the impact of neutralizing antibodies on the evolution of HVR1 was demonstrated in a study by von Hahn and co-workers[57]. Here longitudinal samples were obtained over a time period of 26 years from patient H who was infected in 1977 with genotype 1a. Sera were analyzed for the presence of neutralizing antibodies against the autologous isolate present at the time of sampling. A neutralizing antibody response could be detected as early as 8 wk after infection against the inoculum strain. Interestingly, the antibodies present in a given sample continuously failed to neutralize HCV pseudoparticles bearing the autologous sequence from the same time point. Longitudinal analyses demonstrate continuous escape from emerging antibodies over the time of infection demonstrating humoral immune pressure as the major driving force for the observed sequence diversity in HVR1.
CD8 T cells
Mutational escape from CD8 T cells targeting viral proteins has been well documented for highly variable pathogens such as HIV and SIV. Similarly, in the chimpanzee model of HCV infection selection of mutations in CD8 epitopes that inhibit recognition by specific T cells has been described by Weiner and co-workers[58]. In a follow-up analysis the majority of targeted CD8 epitopes in chimpanzees infected with HCV evolved over time and an important role for mutational escape as a contributor for viral persistence has been suggested[59]. However, acute infection is rarely detected in humans due to lack of specific symptoms making the design of similar longitudinal studies difficult. First evidence for selection pressure by CD8 T cells was obtained from sequence analyses of patients with chronic infection[60]. Here the T cell response against previously defined CD8 epitopes was determined. In some cases the autologous viral sequence present in the patient differed from the described prototype sequence of the epitope and was not targeted by specific T cell lines derived from that patient. The study included a case where sequence evolution was observed in a follow-up sample suggesting that CD8 escape also plays a role during chronic HCV infection. More recently, several longitudinal studies on patients with acute HCV infection have been published providing compelling evidence for CD8 escape in humans[61-64]. Probably the most comprehensive analysis was done by Cox et al[62]. They prospectively followed subjects with ongoing intravenous drug use and high risk behaviour for evidence of acute HCV infection. Using this approach they were able to identify eight patients with acute HCV infection. Samples from these patients were obtained during acute infection (at the time of diagnosis) and after 6 mo. Utilizing comprehensive techniques with overlapping peptides spanning the entire HCV polyprotein the breadth of the immune response was determined. At the same time, sequence evolution between the first and second sample obtained 6 mo later was analyzed. Seventeen of 25 targeted epitopes evolved over time consistent with selection of escape mutations. Of note, the single subject without selection of escape mutations cleared viremia spontaneously. In turn, 50% of the observed sequence changes outside the envelope were associated with a detectable CD8 response. In line with these findings Ray et al analyzed sequences from a single source outbreak infected with HCV genotype 1b and observed reproducible selection of mutations in previously described CD8 epitopes in subjects expressing the restricting HLA-allele[65]. The observation of widespread escape in individuals during acute HCV infection prompted efforts to determine whether adaptation to HLA class I-restricted selection pressure also occurs at the population level. Moore et al analyzed sequences spanning the reverse transcriptase protein of HIV-1 in a large HLA-diverse cohort[66]. This study revealed accumulation of viral sequence polymorphisms at different sites of the protein in patients sharing the same HLA-allele. Many of these sequence polymorphisms were located inside previously described CD8 epitopes that are restricted by the associated HLA-class I allele. This study demonstrated that MHC class I-associated selection pressure has a major impact on the evolution of HIV-1. A similar analysis was done by the same group in a cohort chronically infected with HCV[67]. Here, sequences spanning parts of the NS3 protein were analyzed and again a number of associations between sequence polymorphisms and particular HLA-alleles were identified. This analysis was extended to all non-structural proteins in a cohort of 70 subjects with chronic HCV genotype 1a infection with similar results[68]. An example of an HLA class I-associated sequence polymorphism is illustrated in Figure 2. Viral sequences from all 70 subjects are aligned to a majority consensus sequence and sorted into sequences derived from HLA B8-positive and HLA B8-negative subjects. Boxed is the region of a described HLA B8-restricted CD8 epitope. Differences from the consensus sequence are significantly more frequent in HLA-B8 positive subjects compared to HLA B8-negative subjects indicating that there is reproducible selection pressure on this region in HLA B8-positive subjects. This study included a phylogenetic analysis approach for the detection of HLA-associated sequence polymorphisms highlighting the potential for false positive detection of such associations by pure statistical approaches. However, these studies suggest that the same evolutionary forces act on HCV and HIV-1 and that selection pressure by virus specific CD8 T cells is an important driver of viral evolution.
Figure 2.
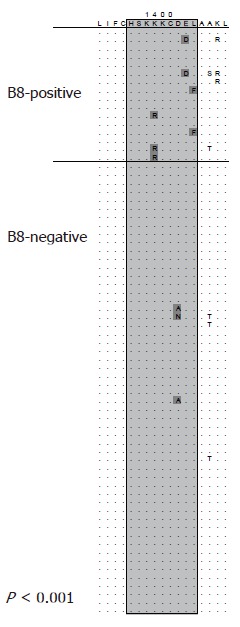
HLA class I-associated sequence polymorphisms in a CD8 epitope. Viral sequences are aligned to a consensus sequence and sorted into sequences derived from HLA B8-positive and HLA B8-negative subjects. Boxed is the region of a described HLA B8-restricted CD8 epitope. Differences from the consensus sequence are significantly more frequent in HLA-B8 positive subjects (P < 0.001). This association was reported in[68].
CD4 T cells
Spontaneous resolution of viremia after acute infection with HCV has been associated with the emergence of a broad and functionally intact T cell response. There is convincing evidence that HCV specific CD4 T cells are important for viral control in the early phase. However, little is known about mutational escape in targeted CD4 T cell epitopes during HCV infection. In theory, a similar selection process as observed for CD8 T cells could be present. A quasispecies that is mutated inside an immuno-dominant CD4 epitope may have a selection advantage in an individual with acute HCV infection and could out compete others. Eckels et al found evidence for selection of mutations in CD4 epitopes in NS3[69,70]. Analysis of a large number of clonal sequences revealed a high degree of polymorphisms in regions targeted by CD4 T cells and the ratio of synonymous versus non-synonymous mutations was consistent with positive selection. However, none of the observed variants became the dominant sequence at a second time point 16 mo later. More recently, two studies have described mutational escape from CD4 responses. The first report is part of a vaccination study in the chimpanzee model[71]. One animal was vaccinated with DNA followed by recombinant vaccinia virus in a prime/boost strategy with HCV NS3 and NS5A/B and subsequently infected with a genotype 1a isolate. After primary infection and transient control of viremia the animal developed chronic infection. Longitudinal sequence analysis of the NS3 and NS5A/B region revealed two non-synonymous mutations. Both of them were located inside regions targeted by CD4 T cells. Additional experiments with synthetic peptides showed that the mutated sequence was not recognized by specific T cells from this animal. A second study focusing on the evolution of the envelope protein in one subject similarly identified amino acid substitutions inside targeted CD4 epitopes consistent with escape[57]. These findings suggest that CD4 T cells may select mutations during HCV infection; however, the overall extent of CD4 escape as a contributor to the evolution of HCV remains to be clarified.
Innate immunity
Infection with HCV initiates a cascade of events within the infected cell with the goal to generate an antiviral state. The first line of defence builds the innate immune system that is triggered by engagement of pathogen-associated molecular patterns (PAMPs) to specific PAMP receptors[72]. In case of HCV toll-like receptor 3 (TLR3) and retinoic-acid-inducible gene I (RIG-I)-receptor recognize dsRNA resulting in activation of multiple cellular factors that mediate transcription and secretion of interferon alpha and beta. Engagement of these type I interferons with their cellular receptor activates a series of interferon stimulated genes (ISG) with the goal to initiate an antiviral state within the infected cell. The hepatitis C virus has developed strategies to evade this first line of host immune defence. Recently it was demonstrated that the NS3/4A protease is able to specifically cleave Cardif (CARD adaptor inducing interferon beta) and TRIF (Toll-interleukin-1 receptor domain-containing adaptor-inducing beta interferon)[73-75]. Both proteins are involved in the activation of interferon regulatory factors (IRFs) and their inactivation, therefore, interferes with the production of interferon alpha and beta. Gale et al[76] demonstrated in vitro that NS5A is able to inhibit protein kinase R (PKR). PKR is activated by interferon alpha and involved in the inhibition of viral RNA translation. Its inhibition, therefore, represents a functional antagonism to the interferon alpha response. Interactions between virus and cellular host factors are sequence specific and negative selection of mutations undermining these immune evasion strategies seems reasonable. However, the underlying mechanisms of the innate immune response are highly conserved in all humans and less dependent on the genetic background of the individual. Therefore, the direction of selection pressure does not change upon transmission to the next host, which makes the observation of positive selection unlikely. Moreover, the interactions between virus and the host’s innate immune response take place during the earliest phase of infection making it even more difficult to directly show that viral escape variants are selected.
Drugs
The current standard treatment regimen for patients with chronic hepatitis C is a combination of pegylated interferon alpha with ribavirin[2,3]. Interestingly, the response rate to this treatment regimen is dependent on the infecting genotype suggesting that sequence differences between genotypes influence the susceptibility to these drugs. Patients infected with genotype 2 and 3 usually show a much faster decline in viral load after initiation of therapy associated with higher sustained response rates. The determinants of this differential responsiveness of different genotypes are poorly understood. Interferon alpha predominantly modulates the immune system[77]. Engagement of its specific receptor turns on a cascade of IFN-stimulated genes (ISGs) resulting in a non-pathogen specific antiviral state. Different HCV sequences seem to have different capabilities to interfere with this anti-viral strategy. The response rates to treatment dramatically differ not only between different genotypes, but also between isolates of the same subtype. Comparison of sequence isolates that have been successfully treated with isolates that did not respond has put a 40 amino acid stretch of the HCV NS5A protein into the spotlight[78]. The degree of sequence variation in this region has been associated with treatment outcome and has therefore been designated as the interferon-sensitivity determining region (ISDR). Subsequently, conflicting results have been published in similar studies; however, a meta-analysis supported the impact of this region on treatment outcome[79]. A correlate of this observation may be the reported inhibitory action of HCV NS5A on PKR[76]. For this interaction the ISDR and an additional 26 C-terminal amino acid stretch of NS5A are crucial. Therefore, selection of viral variants during treatment that successfully enhance this interaction seems reasonable. However, neither was selection of mutations observed in the presence of this antiviral drug in vitro[80] nor has selection of variants during treatment in longitudinal studies formally been shown. It remains, therefore, unclear how interferon alpha contributes to evolution.
The exact antiviral mechanisms of ribavirin are even less well established. Several mechanisms have been suggested including inhibition of the HCV polymerase and early chain termination during the replication process. Higher mutation rates in the presence of ribavirin have been reported potentially resulting in an ‘error catastrophe’. Two recent studies analyzed the mutation rate in the presence and absence of ribavirin in patients receiving treatment. Hofmann et al analyzed the NS3 and NS5B gene in 14 subjects receiving either ribavirin monotherapy or in combination with interferon alpha[81]. Based on a comparison of clonal sequences in the quasispecies population before and after initiation of therapy they concluded that the mutation rate of HCV is higher in the presence of ribavirin. These results were reproducible in cell culture with HCV replicon bearing hepatoma cell lines. Even though the overall effect was weak, a dose dependency could be demonstrated and the inactive L-enantiomer did not show the same effect. In a similar analysis by Lutchman et al the mutation rate for NS5B was calculated based on analysis of bulk and clonal sequences of the NS5B gene in 18 subjects receiving ribavirin and 13 subjects receiving placebo[82]. A significant increase of the mutation rate in the presence of ribavirin was observed after 4 wk of treatment; however, there was no significant difference between the mutation rates after 24 wk compared to the placebo group. The authors of this latter study conclude that ribavirin unlikely acts through an increase of the mutational error rate resulting in an error catastrophe. One study demonstrated selection of a Phe to Tyr mutation in position 415 of the HCV NS5B protein in the presence of ribavirin[83]. This mutation was associated with a less susceptible phenotype for this drug when tested in the replicon model in vitro. This mutation was also observed in 5 out of 16 subjects infected with genotype 1a in the study by Lutchman et al. However, it was not reproducible in HCV genotype 1b. It is, therefore, still unclear if specific mutations are selected in the presence of ribavirin.
Future treatment strategies will include small molecules as inhibitors of virus specific protein functions. Drugs like protease and polymerase inhibitors are very successful for the treatment of HIV. However, selection of variants that are resistant to these antiviral compounds is a major challenge in the management of patients infected with HIV. In recent years many compounds have been tested as inhibitors of the HCV protease and polymerase. Some are now available in early clinical trials. The first compound that was tested in humans was the protease inhibitor BILN 2061. In patients infected with genotype 1 the viral load was dramatically decreased after only 2 d of treatment[84]. However, the drug was designed to inhibit the HCV protease from a genotype 1 isolate with high affinity. As expected, the efficacy was, therefore, much lower in subjects infected with HCV genotype 2 or 3[85]. Due to observed toxic effects of this drug in high dosage in animal models further clinical trials were stopped. Other small compounds have meanwhile reached early phases of clinical testing (reviewed in[86]) including protease inhibitors (VX-950 and SCH 503034) and polymerase inhibitors (NM283 and HCV-796). Along with their proof of excellent efficacy in vitro and in vivo several reports have been published describing resistance mutations[87,88]. A recent study by Sarrazin et al analyzed clonal sequences from subjects treated with the protease inhibitor VX-950 in a clinical trial[89]. In this analysis mutations associated with phenotypic resistance were rapidly selected during treatment and the number of resistant clones in each patient correlated well with the virologic response to the drug. The mutations were reproducibly located in only a few positions in the HCV protease gene. Interestingly, the number of resistant clones decreased after cessation of therapy indicating that some mutations are associated with fitness costs and revert back to wild type in the absence of the drug. Future studies will show if combinations of different drugs such as polymerase and protease inhibitors are beneficial to decrease the risk of resistance mutations similar to HIV.
Constraints on sequence diversity-purifying selection
Analysis of available HCV full genome sequences from public databases shows that the degree of sequence variation varies both between different proteins, but also between regions of the same protein (Figure 3). Some regions are highly conserved even across different HCV genotypes. Many of these highly conserved regions represent functionally important motifs in the viral protein in which substantial sequence variation is not tolerated. Viral evolution is clearly limited by structural constraints forcing the virus into a state in which it is able to functionally exist. Many mutations that occur during the replication process are deleterious or disadvantageous to the fitness of the virus and are, therefore, negatively selected. In contrast, as highlighted in this review multiple forces of the immune system or in some cases drugs exact positive selection pressure away from the consensus sequence in the individual. Selection of variants is, therefore, a trade-off between host pressure and functional needs. Purifying selection describes the driving force towards a sequence with optimal replication capacity in the absence of outside pressure on the virus. Reversion of resistance mutations that have been selected in the presence of antiviral drugs back to the consensus sequence have been first described in the influenza model[90]. For HIV, reversion of drug resistance mutations is well documented and the concept of a salvage therapy was based on this observation. In this concept treatment is re-initiated after interruption and genotypic reversion of drug resistance. However, the clinical benefit is controversial and with a wide range of antiviral drugs now available for the treatment of HIV this strategy received less attention.
Figure 3.
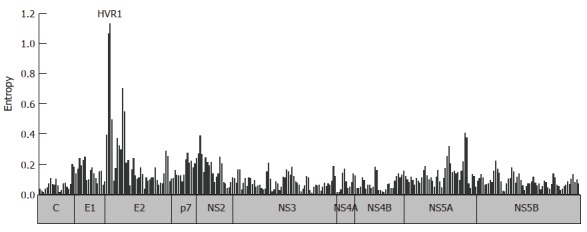
Entropy across the HCV polyprotein. 100 HCV genotype 1b sequences were retrieved from the Los Alamos National Laboratory (LANL) HCV Sequence Database. The entropy score was calculated for 300 windows of 20 residues overlapping by 10 residues utilizing the algorithm implemented in the database.
In HCV reversion was first described for an escape mutation that has been selected by virus-specific CTLs[64]. In this study a virus harbouring an escape mutation in an HLA-B8 restricted epitope in NS3 was transmitted to a host who is HLA-B8 negative and who, therefore, was not able to mount the same T cell response. In the new host the virus continued to evolve back to the prototype sequence and the variant disappeared. Similarly, Ray et al analyzed viral sequences from a single source outbreak years after the transmission event[65]. Interestingly, the source of the virus again had an escape mutation in the same HLA-B8 restricted epitope in NS3. Years later the mutation has reverted back to the prototype sequence in most recipients. Of note, the mutation was stable in subjects who are HLA-B8 positive and who are theoretically able to target this region. Similar to the reversion of CTL escape mutations Sarrazin et al[89]describe in their study on drug resistance to the protease inhibitor VX-960 reversion back to the wild type sequence after treatment was discontinued. These studies illustrate the main selecting forces of HCV evolution. On one side, there is positive selection pressure mainly by the immune system but also in the presence of antiviral drugs. These selection forces are not constant and vary in different hosts and different environments largely depending on the host’s genetic background. On the other side, there is negative selection pressure, which presses the virus into a state of optimal replication capacity. This force is more or less constant, but largely depends on the pre-existing sequence configuration such as the genotype or presence of compensatory mutations.
CONCLUSION
The inherent sequence diversity of HCV represents a major challenge for any treatment. HCV has the ability to rapidly adapt to a hostile environment in the acutely infected individual with an activated immune system at work. An important feature of any therapeutic or prophylactic intervention will be the efficient attack of a structurally or functionally important region in the viral protein. Ideally the attack is targeted against a region that does not tolerate substantial sequence variation in order to limit the ability to evade. Alternatively, a combination of different attacks at different sites may decrease the risk of efficient mutational escape. Recent studies in HIV have suggested that selection of escape mutations may actually be linked to control of viral replication[91]. Indeed, many of the described CTL escape mutations particularly in the presence of protective HLA-alleles such as HLA-B27 and -B57 as well as some drug resistance mutations in HIV are associated with dramatic fitness costs for the virus. For HCV, little is known about the impact of escape and drug resistance mutations on replication capacity even though their documented reversion in the absence of the drug or immune pressure suggests similar fitness costs. With the recent development of culture systems that include the complete replication cycle we now have the tools at hand to address important questions on the interaction between different selecting forces acting on HCV. The understanding of this interaction is fundamental for the design of novel therapies.
Footnotes
Supported by the German Hepatitis Network (BMBF) and the Deutsche Forschungsgemeinschaft
S- Editor Ma N L- Editor Rippe RA E- Editor Ma WH
References
- 1.World Health Organization. Hepatitis C-global prevalence (update) Weekly Epidemiological Record. 1999;74:425. [Google Scholar]
- 2.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Gonçales FL, Häussinger D, Diago M, Carosi G, Dhumeaux D, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 3.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–965. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 4.Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436:933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 5.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 7.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA, Frank J. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science. 2001;291:1959–1962. doi: 10.1126/science.1058409. [DOI] [PubMed] [Google Scholar]
- 9.Lohmann V, Koch JO, Bartenschlager R. Processing pathways of the hepatitis C virus proteins. J Hepatol. 1996;24:11–19. [PubMed] [Google Scholar]
- 10.Liu Q, Tackney C, Bhat RA, Prince AM, Zhang P. Regulated processing of hepatitis C virus core protein is linked to subcellular localization. J Virol. 1997;71:657–662. doi: 10.1128/jvi.71.1.657-662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartosch B, Cosset FL. Cell entry of hepatitis C virus. Virology. 2006;348:1–12. doi: 10.1016/j.virol.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 12.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 13.Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J Virol. 2007;81:374–383. doi: 10.1128/JVI.01134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akazawa D, Date T, Morikawa K, Murayama A, Miyamoto M, Kaga M, Barth H, Baumert TF, Dubuisson J, Wakita T. CD81 expression is important for the permissiveness of Huh7 cell clones for heterogeneous hepatitis C virus infection. J Virol. 2007;81:5036–5045. doi: 10.1128/JVI.01573-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci USA. 2003;100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartosch B, Vitelli A, Granier C, Goujon C, Dubuisson J, Pascale S, Scarselli E, Cortese R, Nicosia A, Cosset FL. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J Biol Chem. 2003;278:41624–41630. doi: 10.1074/jbc.M305289200. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Randall G, Higginbottom A, Monk P, Rice CM, McKeating JA. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J Virol. 2004;78:1448–1455. doi: 10.1128/JVI.78.3.1448-1455.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 19.Pavlović D, Neville DC, Argaud O, Blumberg B, Dwek RA, Fischer WB, Zitzmann N. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc Natl Acad Sci USA. 2003;100:6104–6108. doi: 10.1073/pnas.1031527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffin SD, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, Harris MP, Rowlands DJ. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 2003;535:34–38. doi: 10.1016/s0014-5793(02)03851-6. [DOI] [PubMed] [Google Scholar]
- 21.Frick DN. The hepatitis C virus NS3 protein: a model RNA helicase and potential drug target. Curr Issues Mol Biol. 2007;9:1–20. [PMC free article] [PubMed] [Google Scholar]
- 22.Tellinghuisen TL, Rice CM. Interaction between hepatitis C virus proteins and host cell factors. Curr Opin Microbiol. 2002;5:419–427. doi: 10.1016/s1369-5274(02)00341-7. [DOI] [PubMed] [Google Scholar]
- 23.Xu Z, Choi J, Yen TS, Lu W, Strohecker A, Govindarajan S, Chien D, Selby MJ, Ou J. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J. 2001;20:3840–3848. doi: 10.1093/emboj/20.14.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simmonds P, Alberti A, Alter HJ, Bonino F, Bradley DW, Brechot C, Brouwer JT, Chan SW, Chayama K, Chen DS. A proposed system for the nomenclature of hepatitis C viral genotypes. Hepatology. 1994;19:1321–1324. [PubMed] [Google Scholar]
- 25.Pybus OG, Drummond AJ, Nakano T, Robertson BH, Rambaut A. The epidemiology and iatrogenic transmission of hepatitis C virus in Egypt: a Bayesian coalescent approach. Mol Biol Evol. 2003;20:381–387. doi: 10.1093/molbev/msg043. [DOI] [PubMed] [Google Scholar]
- 26.Ray SC, Arthur RR, Carella A, Bukh J, Thomas DL. Genetic epidemiology of hepatitis C virus throughout egypt. J Infect Dis. 2000;182:698–707. doi: 10.1086/315786. [DOI] [PubMed] [Google Scholar]
- 27.Candotti D, Temple J, Sarkodie F, Allain JP. Frequent recovery and broad genotype 2 diversity characterize hepatitis C virus infection in Ghana, West Africa. J Virol. 2003;77:7914–7923. doi: 10.1128/JVI.77.14.7914-7923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong DA, Tong LK, Lim W. High prevalence of hepatitis C virus genotype 6 among certain risk groups in Hong Kong. Eur J Epidemiol. 1998;14:421–426. doi: 10.1023/a:1007400304726. [DOI] [PubMed] [Google Scholar]
- 29.Hissar SS, Goyal A, Kumar M, Pandey C, Suneetha PV, Sood A, Midha V, Sakhuja P, Malhotra V, Sarin SK. Hepatitis C virus genotype 3 predominates in North and Central India and is associated with significant histopathologic liver disease. J Med Virol. 2006;78:452–458. doi: 10.1002/jmv.20561. [DOI] [PubMed] [Google Scholar]
- 30.Simmonds P, Bukh J, Combet C, Deléage G, Enomoto N, Feinstone S, Halfon P, Inchauspé G, Kuiken C, Maertens G, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 31.Combet C, Garnier N, Charavay C, Grando D, Crisan D, Lopez J, Dehne-Garcia A, Geourjon C, Bettler E, Hulo C, et al. euHCVdb: the European hepatitis C virus database. Nucleic Acids Res. 2007;35:D363–D366. doi: 10.1093/nar/gkl970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuiken C, Yusim K, Boykin L, Richardson R. The Los Alamos hepatitis C sequence database. Bioinformatics. 2005;21:379–384. doi: 10.1093/bioinformatics/bth485. [DOI] [PubMed] [Google Scholar]
- 33.Simmonds P. Genetic diversity and evolution of hepatitis C virus--15 years on. J Gen Virol. 2004;85:3173–3188. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- 34.Drake JW, Charlesworth B, Charlesworth D, Crow JF. Rates of spontaneous mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, Perelson AS. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 36.Quer J, Esteban JI, Cos J, Sauleda S, Ocaña L, Martell M, Otero T, Cubero M, Palou E, Murillo P, et al. Effect of bottlenecking on evolution of the nonstructural protein 3 gene of hepatitis C virus during sexually transmitted acute resolving infection. J Virol. 2005;79:15131–15141. doi: 10.1128/JVI.79.24.15131-15141.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nainan OV, Lu L, Gao FX, Meeks E, Robertson BH, Margolis HS. Selective transmission of hepatitis C virus genotypes and quasispecies in humans and experimentally infected chimpanzees. J Gen Virol. 2006;87:83–91. doi: 10.1099/vir.0.81268-0. [DOI] [PubMed] [Google Scholar]
- 38.Rispeter K, Lu M, Behrens SE, Fumiko C, Yoshida T, Roggendorf M. Hepatitis C virus variability: sequence analysis of an isolate after 10 years of chronic infection. Virus Genes. 2000;21:179–188. doi: 10.1023/a:1008135413215. [DOI] [PubMed] [Google Scholar]
- 39.Smith DB, Simmonds P. Characteristics of nucleotide substitution in the hepatitis C virus genome: constraints on sequence change in coding regions at both ends of the genome. J Mol Evol. 1997;45:238–246. doi: 10.1007/pl00006226. [DOI] [PubMed] [Google Scholar]
- 40.Kimura M. Evolutionary rate at the molecular level. Nature. 1968;217:624–626. doi: 10.1038/217624a0. [DOI] [PubMed] [Google Scholar]
- 41.Cochrane A, Searle B, Hardie A, Robertson R, Delahooke T, Cameron S, Tedder RS, Dusheiko GM, De Lamballerie X, Simmonds P. A genetic analysis of hepatitis C virus transmission between injection drug users. J Infect Dis. 2002;186:1212–1221. doi: 10.1086/344314. [DOI] [PubMed] [Google Scholar]
- 42.Pybus OG, Charleston MA, Gupta S, Rambaut A, Holmes EC, Harvey PH. The epidemic behavior of the hepatitis C virus. Science. 2001;292:2323–2325. doi: 10.1126/science.1058321. [DOI] [PubMed] [Google Scholar]
- 43.de Oliveira T, Pybus OG, Rambaut A, Salemi M, Cassol S, Ciccozzi M, Rezza G, Gattinara GC, D'Arrigo R, Amicosante M, et al. Molecular epidemiology: HIV-1 and HCV sequences from Libyan outbreak. Nature. 2006;444:836–837. doi: 10.1038/444836a. [DOI] [PubMed] [Google Scholar]
- 44.Allain JP, Dong Y, Vandamme AM, Moulton V, Salemi M. Evolutionary rate and genetic drift of hepatitis C virus are not correlated with the host immune response: studies of infected donor-recipient clusters. J Virol. 2000;74:2541–2549. doi: 10.1128/jvi.74.6.2541-2549.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grenfell BT, Pybus OG, Gog JR, Wood JL, Daly JM, Mumford JA, Holmes EC. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–332. doi: 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- 46.Farci P, Shimoda A, Coiana A, Diaz G, Peddis G, Melpolder JC, Strazzera A, Chien DY, Munoz SJ, Balestrieri A, et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science. 2000;288:339–344. doi: 10.1126/science.288.5464.339. [DOI] [PubMed] [Google Scholar]
- 47.Farci P, Strazzera R, Alter HJ, Farci S, Degioannis D, Coiana A, Peddis G, Usai F, Serra G, Chessa L, et al. Early changes in hepatitis C viral quasispecies during interferon therapy predict the therapeutic outcome. Proc Natl Acad Sci USA. 2002;99:3081–3086. doi: 10.1073/pnas.052712599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chambers TJ, Fan X, Droll DA, Hembrador E, Slater T, Nickells MW, Dustin LB, Dibisceglie AM. Quasispecies heterogeneity within the E1/E2 region as a pretreatment variable during pegylated interferon therapy of chronic hepatitis C virus infection. J Virol. 2005;79:3071–3083. doi: 10.1128/JVI.79.5.3071-3083.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kato N, Ootsuyama Y, Sekiya H, Ohkoshi S, Nakazawa T, Hijikata M, Shimotohno K. Genetic drift in hypervariable region 1 of the viral genome in persistent hepatitis C virus infection. J Virol. 1994;68:4776–4784. doi: 10.1128/jvi.68.8.4776-4784.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiner AJ, Geysen HM, Christopherson C, Hall JE, Mason TJ, Saracco G, Bonino F, Crawford K, Marion CD, Crawford KA. Evidence for immune selection of hepatitis C virus (HCV) putative envelope glycoprotein variants: potential role in chronic HCV infections. Proc Natl Acad Sci USA. 1992;89:3468–3472. doi: 10.1073/pnas.89.8.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Booth JC, Kumar U, Webster D, Monjardino J, Thomas HC. Comparison of the rate of sequence variation in the hypervariable region of E2/NS1 region of hepatitis C virus in normal and hypogammaglobulinemic patients. Hepatology. 1998;27:223–227. doi: 10.1002/hep.510270134. [DOI] [PubMed] [Google Scholar]
- 52.Christie JM, Chapel H, Chapman RW, Rosenberg WM. Immune selection and genetic sequence variation in core and envelope regions of hepatitis C virus. Hepatology. 1999;30:1037–1044. doi: 10.1002/hep.510300403. [DOI] [PubMed] [Google Scholar]
- 53.Ray SC, Mao Q, Lanford RE, Bassett S, Laeyendecker O, Wang YM, Thomas DL. Hypervariable region 1 sequence stability during hepatitis C virus replication in chimpanzees. J Virol. 2000;74:3058–3066. doi: 10.1128/jvi.74.7.3058-3066.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Logvinoff C, Major ME, Oldach D, Heyward S, Talal A, Balfe P, Feinstone SM, Alter H, Rice CM, McKeating JA. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc Natl Acad Sci USA. 2004;101:10149–10154. doi: 10.1073/pnas.0403519101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drummer HE, Maerz A, Poumbourios P. Cell surface expression of functional hepatitis C virus E1 and E2 glycoproteins. FEBS Lett. 2003;546:385–390. doi: 10.1016/s0014-5793(03)00635-5. [DOI] [PubMed] [Google Scholar]
- 56.Bartosch B, Bukh J, Meunier JC, Granier C, Engle RE, Blackwelder WC, Emerson SU, Cosset FL, Purcell RH. In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc Natl Acad Sci USA. 2003;100:14199–14204. doi: 10.1073/pnas.2335981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.von Hahn T, Yoon JC, Alter H, Rice CM, Rehermann B, Balfe P, McKeating JA. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology. 2007;132:667–678. doi: 10.1053/j.gastro.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 58.Weiner A, Erickson AL, Kansopon J, Crawford K, Muchmore E, Hughes AL, Houghton M, Walker CM. Persistent hepatitis C virus infection in a chimpanzee is associated with emergence of a cytotoxic T lymphocyte escape variant. Proc Natl Acad Sci USA. 1995;92:2755–2759. doi: 10.1073/pnas.92.7.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J, McKinney D, Sette A, Hughes AL, Walker CM. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity. 2001;15:883–895. doi: 10.1016/s1074-7613(01)00245-x. [DOI] [PubMed] [Google Scholar]
- 60.Chang KM, Rehermann B, McHutchison JG, Pasquinelli C, Southwood S, Sette A, Chisari FV. Immunological significance of cytotoxic T lymphocyte epitope variants in patients chronically infected by the hepatitis C virus. J Clin Invest. 1997;100:2376–2385. doi: 10.1172/JCI119778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guglietta S, Garbuglia AR, Pacciani V, Scottà C, Perrone MP, Laurenti L, Spada E, Mele A, Capobianchi MR, Taliani G, et al. Positive selection of cytotoxic T lymphocyte escape variants during acute hepatitis C virus infection. Eur J Immunol. 2005;35:2627–2637. doi: 10.1002/eji.200526067. [DOI] [PubMed] [Google Scholar]
- 62.Cox AL, Mosbruger T, Mao Q, Liu Z, Wang XH, Yang HC, Sidney J, Sette A, Pardoll D, Thomas DL, et al. Cellular immune selection with hepatitis C virus persistence in humans. J Exp Med. 2005;201:1741–1752. doi: 10.1084/jem.20050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tester I, Smyk-Pearson S, Wang P, Wertheimer A, Yao E, Lewinsohn DM, Tavis JE, Rosen HR. Immune evasion versus recovery after acute hepatitis C virus infection from a shared source. J Exp Med. 2005;201:1725–1731. doi: 10.1084/jem.20042284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Timm J, Lauer GM, Kavanagh DG, Sheridan I, Kim AY, Lucas M, Pillay T, Ouchi K, Reyor LL, Schulze zur Wiesch J, et al. CD8 epitope escape and reversion in acute HCV infection. J Exp Med. 2004;200:1593–1604. doi: 10.1084/jem.20041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ray SC, Fanning L, Wang XH, Netski DM, Kenny-Walsh E, Thomas DL. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J Exp Med. 2005;201:1753–1759. doi: 10.1084/jem.20050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moore CB, John M, James IR, Christiansen FT, Witt CS, Mallal SA. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science. 2002;296:1439–1443. doi: 10.1126/science.1069660. [DOI] [PubMed] [Google Scholar]
- 67.Gaudieri S, Rauch A, Park LP, Freitas E, Herrmann S, Jeffrey G, Cheng W, Pfafferott K, Naidoo K, Chapman R, et al. Evidence of viral adaptation to HLA class I-restricted immune pressure in chronic hepatitis C virus infection. J Virol. 2006;80:11094–11104. doi: 10.1128/JVI.00912-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Timm J, Li B, Daniels MG, Bhattacharya T, Reyor LL, Allgaier R, Kuntzen T, Fischer W, Nolan BE, Duncan J, et al. Human leukocyte antigen-associated sequence polymorphisms in hepatitis C virus reveal reproducible immune responses and constraints on viral evolution. Hepatology. 2007;46:339–349. doi: 10.1002/hep.21702. [DOI] [PubMed] [Google Scholar]
- 69.Eckels DD, Zhou H, Bian TH, Wang H. Identification of antigenic escape variants in an immunodominant epitope of hepatitis C virus. Int Immunol. 1999;11:577–583. doi: 10.1093/intimm/11.4.577. [DOI] [PubMed] [Google Scholar]
- 70.Wang H, Eckels DD. Mutations in immunodominant T cell epitopes derived from the nonstructural 3 protein of hepatitis C virus have the potential for generating escape variants that may have important consequences for T cell recognition. J Immunol. 1999;162:4177–4183. [PubMed] [Google Scholar]
- 71.Puig M, Mihalik K, Tilton JC, Williams O, Merchlinsky M, Connors M, Feinstone SM, Major ME. CD4+ immune escape and subsequent T-cell failure following chimpanzee immunization against hepatitis C virus. Hepatology. 2006;44:736–745. doi: 10.1002/hep.21319. [DOI] [PubMed] [Google Scholar]
- 72.Gale M, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436:939–945. doi: 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- 73.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Lemon SM. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Breiman A, Grandvaux N, Lin R, Ottone C, Akira S, Yoneyama M, Fujita T, Hiscott J, Meurs EF. Inhibition of RIG-I-dependent signaling to the interferon pathway during hepatitis C virus expression and restoration of signaling by IKKepsilon. J Virol. 2005;79:3969–3978. doi: 10.1128/JVI.79.7.3969-3978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 76.Gale M, Blakely CM, Kwieciszewski B, Tan SL, Dossett M, Tang NM, Korth MJ, Polyak SJ, Gretch DR, Katze MG. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol Cell Biol. 1998;18:5208–5218. doi: 10.1128/mcb.18.9.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 78.Chayama K, Tsubota A, Kobayashi M, Okamoto K, Hashimoto M, Miyano Y, Koike H, Kobayashi M, Koida I, Arase Y, et al. Pretreatment virus load and multiple amino acid substitutions in the interferon sensitivity-determining region predict the outcome of interferon treatment in patients with chronic genotype 1b hepatitis C virus infection. Hepatology. 1997;25:745–749. doi: 10.1002/hep.510250342. [DOI] [PubMed] [Google Scholar]
- 79.Pascu M, Martus P, Höhne M, Wiedenmann B, Hopf U, Schreier E, Berg T. Sustained virological response in hepatitis C virus type 1b infected patients is predicted by the number of mutations within the NS5A-ISDR: a meta-analysis focused on geographical differences. Gut. 2004;53:1345–1351. doi: 10.1136/gut.2003.031336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aus dem Siepen M, Oniangue-Ndza C, Wiese M, Ross S, Roggendorf M, Viazov S. Interferon-alpha and ribavirin resistance of Huh7 cells transfected with HCV subgenomic replicon. Virus Res. 2007;125:109–113. doi: 10.1016/j.virusres.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 81.Hofmann WP, Polta A, Herrmann E, Mihm U, Kronenberger B, Sonntag T, Lohmann V, Schönberger B, Zeuzem S, Sarrazin C. Mutagenic effect of ribavirin on hepatitis C nonstructural 5B quasispecies in vitro and during antiviral therapy. Gastroenterology. 2007;132:921–930. doi: 10.1053/j.gastro.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 82.Lutchman G, Danehower S, Song BC, Liang TJ, Hoofnagle JH, Thomson M, Ghany MG. Mutation rate of the hepatitis C virus NS5B in patients undergoing treatment with ribavirin monotherapy. Gastroenterology. 2007;132:1757–1766. doi: 10.1053/j.gastro.2007.03.035. [DOI] [PubMed] [Google Scholar]
- 83.Young KC, Lindsay KL, Lee KJ, Liu WC, He JW, Milstein SL, Lai MM. Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology. 2003;38:869–878. doi: 10.1053/jhep.2003.50445. [DOI] [PubMed] [Google Scholar]
- 84.Hinrichsen H, Benhamou Y, Wedemeyer H, Reiser M, Sentjens RE, Calleja JL, Forns X, Erhardt A, Crönlein J, Chaves RL, et al. Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology. 2004;127:1347–1355. doi: 10.1053/j.gastro.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 85.Reiser M, Hinrichsen H, Benhamou Y, Reesink HW, Wedemeyer H, Avendano C, Riba N, Yong CL, Nehmiz G, Steinmann GG. Antiviral efficacy of NS3-serine protease inhibitor BILN-2061 in patients with chronic genotype 2 and 3 hepatitis C. Hepatology. 2005;41:832–835. doi: 10.1002/hep.20612. [DOI] [PubMed] [Google Scholar]
- 86.Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology. 2007;132:1979–1998. doi: 10.1053/j.gastro.2007.03.116. [DOI] [PubMed] [Google Scholar]
- 87.Lin C, Lin K, Luong YP, Rao BG, Wei YY, Brennan DL, Fulghum JR, Hsiao HM, Ma S, Maxwell JP, et al. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: structural analysis indicates different resistance mechanisms. J Biol Chem. 2004;279:17508–17514. doi: 10.1074/jbc.M313020200. [DOI] [PubMed] [Google Scholar]
- 88.Tong X, Chase R, Skelton A, Chen T, Wright-Minogue J, Malcolm BA. Identification and analysis of fitness of resistance mutations against the HCV protease inhibitor SCH 503034. Antiviral Res. 2006;70:28–38. doi: 10.1016/j.antiviral.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 89.Sarrazin C, Kieffer TL, Bartels D, Hanzelka B, Müh U, Welker M, Wincheringer D, Zhou Y, Chu HM, Lin C, et al. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology. 2007;132:1767–1777. doi: 10.1053/j.gastro.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 90.Grambas S, Bennett MS, Hay AJ. Influence of amantadine resistance mutations on the pH regulatory function of the M2 protein of influenza A viruses. Virology. 1992;191:541–549. doi: 10.1016/0042-6822(92)90229-i. [DOI] [PubMed] [Google Scholar]
- 91.Altfeld M, Allen TM. Hitting HIV where it hurts: an alternative approach to HIV vaccine design. Trends Immunol. 2006;27:504–510. doi: 10.1016/j.it.2006.09.007. [DOI] [PubMed] [Google Scholar]