

Abstract
AIM: To investigate the hotspots, direction, and the time course of evolution of hepatitis A virus in the process of consecutive cell culture passage in human KMB17 diploid cells.
METHODS: Wild type hepatitis A virus H2w was serially propagated in KMB17 cells until passage 30, and the full-length genomes of H2w and its six chosen progenies were determined by directly sequencing RT-PCR products amplified from viral genomic RNA. Alignment comparison of sequences from H2w with its six progenies and phylogenetic analysis of the whole VP1 region from H2w, progenies of H2w, and other cell culture adapted hepatitis A virus were then carried out to obtain data on the molecular evolution of hepatitis A virus in the process of consecutive passage in KMB17 cells.
RESULTS: Most of the mutations occurred by passage 5 and several hotspots related to adaptation of the virus during cell growth were observed. After that stage, few additional mutations occurred through the remaining duration of passage in KMB17 cells except for mutation in the virulence determinants, which occurred in the vicinity of passage 15. The phylogenetic analysis of the whole VP1 region suggested that the progenies of H2w evolved closely to other cell culture adapted hepatitis A virus, i.e. MBB, L-A-1, other than its progenitor H2w.
CONCLUSION: Hepatitis A virus served as a useful model for studying molecular evolution of viruses in a given environment. The information obtained in this study may provide assistance in cultivating the next generation of a seed virus for live hepatitis A vaccine production.
Keywords: Molecular evolution, Virus evolution, Phylogeny analysis, Virulence gene, Hepatitis A virus
INTRODUCTION
The mechanism of molecular evolution is often used by epidemiologists to acquire information about the direction of an epidemic, velocity of dissemination, site of the origin of the epidemic, and even predictions concerning future epidemics[1,2]. These methods are helpful in visualizing molecular events of viruses that have evolved in a known environment, i.e. consecutive propagation of viruses in a given cell line under given temperature and nutritional conditions. However, only a few studies had been carried out to fulfill this objective. Research of this type can provide virologists important information on the evolutionary history of viruses, as well as the virulent gene in the virus.
Our group has accumulated several years of technical expertise in the cultivation of hepatitis A virus (HAV) in the human KMB17 diploid cell line, which originated from human embryonic lung tissue[3]. The aim of the present study was to employ HAV as a model virus to investigate the evolution of the virus in the course of consecutive cell culture passage in human KMB17 diploid cells. In the present study, we serially propagated wild type HAV H2w in KMB17 cells to obtain 30 consecutive progenies, applied cold adaptation at passage 15 to 20 to investigate the conditions resulting in mutation within virulence determinants, and acquired virus samples at the end of every 5 passages from serial progenies for genome-wide sequence analysis. The resulting 5th (H2K5), 10th (H2K10), 15th (H2K15), 20th (H2K20), 25th (H2K25), and 30th (H2K30) progenies were obtained and analyzed. We have attempted to summarize the time course of molecular events that occur in the process of consecutive passage of HAV in human KMB17 diploid cells, as well as the hotspots and the direction of the evolution of HAV in given conditions. We hope that the information obtained from this study will help explain the conditions which promote the development of mutations pertaining to viral virulence determinants, and provide help in the development of a seed virus for the production of live virus vaccine.
MATERIALS AND METHODS
Cells and viruses
The KMB17 cell line which originated from human embryonic lung[4] is now widely used for vaccine production in China[3]. The KMB17 cells were grown in Dulbecco's modified Eagle's medium (Invitrogen), supplemented with 10% newborn calf serum (Invitrogen).
The H2w strain of HAV was isolated directly from feces obtained from hepatitis A patients[5]. The KMB17 cells-adapted viruses were retrieved from cell substrate as follows: cells were first suspended in 6 mL phosphate saline buffer (pH 7.0) and treated with three cycles of freeze-thawing/sonication. The supernatant was obtained after centrifugation at 8000 r/min for 30 min and then extracted three times with chloroform. The virus in the supernatant was precipitated by the addition of polyethylene glycol 6000 to obtain a final concentration of 10% (wt/vol) and sodium chloride to give 0.4 mol/L solution[6]. The virus pellet was collected by centrifugation at 12 000 r/min for 30 min and was used directly in HAV RNA extraction.
Consecutive passage of HAV in KMB17
105 50% tissue culture infectious doses (TCID50) of H2w were inoculated into 80% confluent monolayers of KMB17 cells in a 25 cm2 flask (Costar). After 2 h of adsorption, the cells were washed three times and incubated at 35°C for 45 d to fulfill the first passage in the cell line[3]. When harvested, the cells were suspended in 6 mL phosphate saline buffer (pH 7.0) and treated as described in the “cells and viruses” section above to obtain the 1st progeny virus in the supernatant. One ml of the supernatant was then inoculated into KMB17 cells to perform the 2nd passage. In this passage, the cells were kept at 35°C for 28 d. Consecutive passages from 3rd to 30th passage were carried out using the same technique as for the 2nd passage. All the passages were carried out at 35°C except for six passages from 15th to 20th, which were performed at 32°C.
Design of PCR primers
The HAV genome was divided into ten overlapping fragments, the size of about 1000 bp, and primers specific to both ends of the fragments were designed. To facilitate the process of sequencing and to ensure reproducibility of the results, we added a universal adapter from T7 promoter primer or M13 reverse sequencing primer to the routine primers used in specific amplification of HAV genome (Table 1). Our design of PCR primers allowed us to sequence PCR products with universal primers, which ensured maintaining the same reaction system for sequencing different PCR fragments, thus providing more reproducible results.
Table 1.
Primers used in amplifying HAV genome
| Name | Sequences | Position |
| F1 | T71-TTTCCGGAGYCCCTCTTG | 22-39 |
| R1 | M13R2-ATCTGCCAGAGACAGGA | 806-790 |
| F2 | T7-GAGGTACTCAGGGGCATTTA | 690-709 |
| R2 | M13R-GGATCCAAAGCAAAAGACA | 1567-1549 |
| F3 | T7-AGATTTGGAGTTGCATGGATTAAC | 1430-1453 |
| R3 | M13R-GGATCCTGAAACATCCA | 2342-2326 |
| F4 | M13R-TTCAACAACAGTTTCTACAG | 2231-2250 |
| R4 | T7-AGCTTCCTCCTCTGATCTA | 3098-3080 |
| F5 | T7-GGGAGATAAGACAGATTCTACAT | 2861-2883 |
| R5 | M13R-AGCTTCCACTCTTAACAAA | 3813-3795 |
| F6 | M13R-GAATGCTTGGATTGTCTGG | 3634-3652 |
| R6 | T7-GATCCATCCCAGTAATCTGA | 4549-4530 |
| F7 | T7-ATCAATTGCATTGGCAAC | 4451-4468 |
| R7 | M13R-GGATCCGCATCTAATTTAATC | 5278-5258 |
| F8 | M13R-GCTGTGGGAGCTGCAGTTGG | 5124-5143 |
| R8 | T7-GATCCATGGGACTCTTTCTA | 6027-6008 |
| F9 | T7-GAGGAAATTCAATTCTTG | 5872-5889 |
| R9 | M13R-GGATGGAGTTCCAGA | 6764-6750 |
| F10 | T7-GGAGATGTYGGTCTTGAT | 6657-6674 |
| R10 | M13R-AGAGGGGTCTCCGGGAATTT | polyA |
| 3'RACE | CAAGAGGGGTCTCCGGGAATTTTTTTTTTTTTTTTTTTT | polyA |
T7 indicates adapter with sequence: TAATACGACTCACTATAGGGAGA,
M13R indicates adapter with sequence: AGCGGATAACAATTTCACACAGGA.
Nucleotide sequence analysis
HAV RNA was extracted from virus pellets using Trizol (Promega) according to the manufacturer’s instruction. cDNA was synthesized using primers complementary to HAV nt 1567 to 1549, 3098 to 3080, 4549 to 4530, 6027 to 6008 or 3' RACE primer with AMV reverse transcriptase (Promega). PCR amplifications were performed using 10 pairs of primers specific for HAV genome (Table 1) with Taq polymerase (Promega) based on the routine operation of PCR amplification. Sequences of PCR fragments were determined by directly sequencing the gel-purified PCR fragments on an ABI Prism 3730XL automated sequencer using T7 Promoter primer or M13 reverse sequencing primer. At least three reactions of the same PCR fragment were performed to ensure reliable results. The nucleotide sequence of the full-length HAV genome was obtained by splicing sequences from 10 overlapping RT-PCR fragments. Pair wise comparison of sequences from H2w and its six progenies was performed with Lasergene software package. The number of mutations in the nucleotide occurring in every 5 passages was calculated, and the mutation events as well as mutations in the deduced amino acid were summarized to provide information on hotspots and the time course of mutations of HAV during consecutive cell culture passages.
Subgenotype analysis of progeny viruses
Subgenotype analysis was performed by pair wise comparison of sequences from 168 bp VP1/2A junction at nt 3024 to nt 3191 according to the method of Robertson et al[7]. HM175 and LA were used as reference strains for IB and IA subgenotypes respectively. Divergence less than 7.5% in comparison with the reference strain was used as a criterion for distinguishing different subgenotype.
Phylogenetic assay of cell culture derivatives of H2w with other cell culture adapted HAV
Phylogenetic and molecular evolutionary analyses were conducted to evaluate the direction of evolution of HAV in consecutive passages in KMB17 cells. According to the method described by Costa-Mattioli et al[8], the entire VP1 nucleotide sequences of H2w and its chosen progenies were aligned to other cell culture adapted HAV or reference strains, LA/wt[9], GBM/wt, GBM/FRhK, GBM/HFS[10], HM175/wt[11], HM175/HAV7[12], HM175/18F, HM175/24A, HM175/43C[13], L-A-1[14] and MBB[15], by the CLUSTAL W program[16]. Matrix distances for the Kimura two-parameter model were calculated from the resulting alignment output[17] and used to plot neighbor-joining phylogenetic trees. These methods were implemented by using MEGA software package version 3.1[18].
GenBank accession numbers
Sequences of several HAV strains were drawn from the GenBank for phylogenetic analysis. These included LA/wt (K02990), GBM/wt (X75215), GBM/FRhK (X75214), GBM/HFS (X75216), HM175/wt (M14707), HM175/HAV7 (M16632), HM175/18F (M59808), HM175/24A (M59810), HM175/43C (M59809), L-A-1 (AF314208) and MBB (M20273).
RESULTS
Nucleotide sequences of full-length HAV genomes
Our design allowed us to sequence the PCR products with a high degree of efficiency and reproducibility. All the three reactions of the same PCR fragment gave the same result, with excellent readability. The individual sequences from PCR fragments were spliced to obtain seven nearly full-length genomic sequences, which were deposited in GenBank with accession numbers: H2w (EF406357), H2K5 (EF406358), H2K10 (EF406359), H2K15 (EF406360), H2K20 (EF406361), H2K25 (EF406362) and H2K30 (EF406363).
Conversion of subgenotype during cell culture-adaptation
After pair wise comparison of H2w and its six cell culture derived progenies with HM-175 and LA, a divergence 3.6% of progenies to reference IB strain (HM175) other than 10.1% of the parent H2w was obtained. Often divergence less than 7.5% to the reference strain was a criterion for distinguishing different subgenotype, therefore, cell culture-adaptation switched the subgenotype of progenies from 1A to 1B (Table 2), which was not a common phenomenon in cell culture study of HAV conducted by other workers[12,19].
Table 2.
Divergence of H2w and its progenies compared to reference strains in VP1/2A region (%)
| H2w | H2K5 | H2K10 | H2K15 | K2K20 | H2K25 | H2K30 | |
| HM175 | 10.1 | 3.6 | 3.6 | 3.6 | 3.6 | 3.6 | 3.6 |
| LA | 4.8 | 7.7 | 7.7 | 7.7 | 7.7 | 7.7 | 7.7 |
Hotspot mutations in the course of consecutive cell passage
After alignment comparison of nearly full-length genomic sequences from H2w and its six cell culture derived progenies, we summarized hotspot mutations accumulated in the course of every 5 passages in the KMB17 cell line (Figure 1). Within the first 5 passages, 648 mutations occurred throughout the genome, out of which forty were amino acid mutations. Among these 648 mutations, 9 nucleotides deletion at nt 128 to 136, 11 nucleotide insert at nt 176 to 177, 6 nucleotide deletion at nt 5018 to 5023, U/G mutation at nt 687 and C/U mutation at nt 3889, are possible hotspot mutations common to cell culture adapted HAV, or specific to HAV when adapted to grow in the KMB17 cell line[12,19], which played crucial role in ensuring that the virus grew efficiently in human cell line KMB17. In the course of the next 5 passages, there was only one nucleotide deletion at nt 203, which was common to cell culture adapted HAV as described by other authors[12]. A U/C mutation at nt 4222 occurred during the third 5 passages, which has been shown to be a virulence-related mutation[20]. In the remaining consecutive passages, no new mutations were observed.
Figure 1.
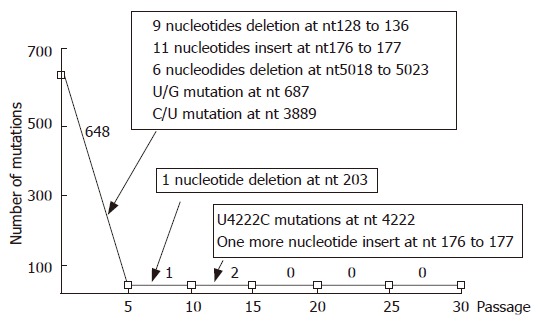
Summary of hotspots and time course of mutations in the process of consecutive passage of HAV in human cell line KMB17. The numbers on the curve indicate total mutations occurring at every 5 passages. Text in the boxes describes hot mutations occurring at the stage of cell culture passage.
Time course of mutations pertaining to cell culture adaptation
Cell culture of HAV has been studied for many years. Several authors have reported their findings on hotspots related to changes in the HAV genome after cell-culture adaptation in different cell lines[12,19,21-25]. Among these findings, C/U mutation at nt 3889 of 2B region[23] was the most important in cell culture adaptation of HAV, regardless of the cell line origin. Other events, such as insertion mutation in 5' NCR and deletions at the beginning of the 3A region, have been suggested as cell line specific phenomena[19,25]. In the present study, we found that almost all the events related to the adaptation of HAV in cell culture appeared to have been completed as early as passage 5 (Figure 1). These events included deletions near nt 131, insert at nt 176 to 177, U/G mutation at nt 687, C/U mutation at nt 3889 along with deletions near nt 5020. All the new mutations were stable during the remainder of the consecutive passage in KMB17 cells. Another mutation common in cell culture adapted HAV[12], was deletion of 1 nucleotide at nt 203, which occurred before passage 10.
Time course of mutations within virulence determinants
It is difficult to identify mutations responsible for the attenuation of HAV. Several studies have examined the virulence determinants of HAV through chimeric viruses constructed from wt HM175 and an attenuated derivative HAV/7[20,26,27]. According to the studies conducted by Emerson et al[20], six nucleotides (HAV nt 3025, nt 3196, nt 4043, nt 4087, nt 4222, nt 4563) may be involved in the attenuation of HAV during cell culture adaptation. When we compared H2w and its cell culture derivatives, only nt 4222 of the six nucleotides mentioned above was found to have mutated from U to C in the long term consecutive cell culture adaptation. We thus extrapolated that in our study, nt 4222 was the main determinant for the virulence of HAV. In present study, we carefully compared H2w and its six chosen derivatives from cell culture at nt 4222, and summarized the time course of mutations related to virulence determinants of HAV. The virulence determinant did not mutate before the 10th passage in KMB17 cell line. Mutation of virulence determinant occurred between passage 11 and passage 15, and these mutations were stable throughout the remainder of the consecutive passage in KMB17 cell line (Figure 1).
Direction of evolution of HAV in prolonged cell culture passage
The complete VP1 nucleotide sequences from H2w and its six progenies were determined and aligned with those of cell culture adapted HAV strains taken from the GenBank. A phylogenetic tree was produced using Kimura two-parameter distance and the neighbor-joining method (Figure 2), which was implemented using the MEGA software package. The results showed that the six progenies of H2w were clustered more closely to MBB, a HAV adapted to human hepatocellular carcinoma cell line[15], and L-A-1, another live attenuated HAV developed in the human diploid cells[14], whereas the progenitor H2w clustered in another group with LA, the reference strain of HAV IA subgenotype. The situation was very different when derivative viruses from HM175 and GBM were examined. The derivatives from HM175 or GBM were clustered closely only with their progenitor virus. The phylogenetic tree suggested that progenies of H2w evolved closely to other cell culture adapted HAV rather than its progenitor virus regardless of the origin of the wild progenitor or the method used for adapting the virus.
Figure 2.
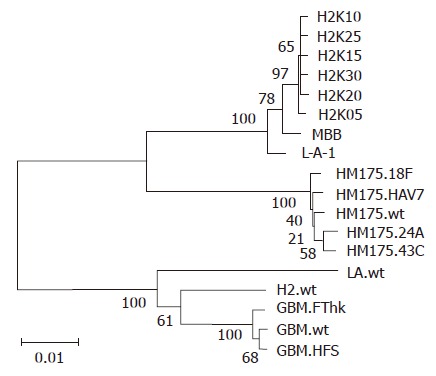
Phylogenetic analysis of the complete VP1 region using MEGA software package version 3.1. The numbers at nodes indicate bootstrap percentages after 1000 replications of bootstrap sampling. The bar indicates genetic distance.
DISCUSSION
In the present study, we used HAV as a model virus to investigate the molecular evolution of HAV in the course of prolonged consecutive passage in KMB17 cells. We obtained a good understanding of the overall time course of molecular events that occur during prolonged consecutive cell culture passage by genome-wide sequence comparison of HAV selected from serial cell culture-derived progenies. After assessing the data on the hotspot mutations at different stages of the consecutive cell culture passage, we concluded that nearly all the mutations occurred before passage 5 and these mutations remained stable throughout the remainder of the prolonged passage in KMB17 cells; several of the hotspot mutations played crucial roles in determining the ability of HAV to grow in cells other than hepatocytes. After this stage, no new mutations were observed except for mutations in the virulence determinants during the remainder of the consecutive passage. If the velocity of mutation is taken into account, our results were different from those described by other workers. Graff et al[19] noted that the GBM strain did not mutate its 5' non-coding region (NCR) before the 8th passage in a human cell line, and before the 20th passage in a rhesus kidney derived cell line. The main factors influencing the velocity of mutation remain unclear. Perhaps the cell substrate plays a critical role in determining the process of mutation. As the phylogenetic analysis of the whole VP1 region from H2w and its derivative progenies with other cell culture derived HAV indicates, cell culture derived progenies of H2w evolve closely to other cell culture derived HAV, i.e., MBB and L-A-1, which were successfully adapted to grow in human cells, despite the origin of the wild type progenitor or the method for adapting the virus. It is possible that the determinant factor for the direction of the evolution may be the cell substrate used, since MBB, L-A-1 and progenies of H2w were all produced in cell lines originating from human tissue.
Only a few studies have examined the time course of mutations within the virulence determinants of HAV, as well as the conditions for the occurrence of the mutations. The information obtained from this evolution study should help explain when and why the virus needed to mutate its virulence gene. In the present study, a U/C mutation at nt 4222 of 2C region with Phe/Ser conversion in amino acid occurred in H2K15, which was crucial for the attenuation of HAV, according to the identification of the virulent gene by Emerson et al[20]. Therefore, HAV may evolve its main virulence determinants in the vicinity of passage 15 in KMB17 cells. Cold adaptation was a common technique used to attenuate the virus[28]. We therefore, hoped that this technique would mutate the virulence determinants of HAV. In our study, after the passage of virus was repeated 14 times at 35°C, we lowered the temperature from 35°C to 32°C for the following 6 passages in order to mutate the virulent gene. The temperature was returned to the normal 35°C after completing the 20th passage so as to obtain the mutants accustomed to the higher temperature. Because U/C mutation at nt 4222 had occurred in H2K15, which was produced at lower temperature, we presumed that this mutation was temperature related. Recently, we sequenced a fragment of H2K14, the 14th progeny virus produced at the normal 35°C, and discovered that the U/C mutation at nt 4222 had already occurred (Data not shown). Therefore, cold adaptation may not influence the mutational process of virulence determinants of HAV. Perhaps, prolonged cell culture adaptation in an appropriate cell line is the real determinant for the occurrence of mutations of the virulent gene. By contrast, a study by Konduru et al[29] showed that the wild type HM175 displayed no mutations during 9 passages in human hepatoma Huh7 cells.
Genetic stability is a major concern in the development of any live viral vaccine. Because of the quasispecies nature of the RNA viruses, it is believed that these viruses are heterogenous in their genome and mutations occur nonspecifically across the genome. In the case of live hepatitis A vaccine, few systematic studies have been carried out to determine the genetic stability of cell culture adapted HAV genome. In the present study, we investigated the genetic stability of cell culture adapted HAV genome after careful observation of the molecular evolution of HAV during prolonged consecutive passage in KBM17 cells. To ascertain that there would be few genetic changes during consecutive passages in KMB17 cells after completion of mutations regarding cell culture adaptation and virulence, we performed alignment analysis of genome-wide sequences from H2w and its progenies resulting from consecutive cell passage. Our results showed that no mutation occurred throughout the whole genome and none of the virulence determinants were affected by prolonged passage in KMB17 cells after the 15th passage. We concluded that the cell culture adapted genome of HAV was very stable during the course of consecutive cell culture passage in human diploid cells.
The data obtained from analyzing the molecular events during prolonged consecutive passage of HAV in KBM17 cells may provide information and technical assistance in monitoring the genetic stability of virulence determinant of seed virus used in live vaccine production, as well as the genetic stability of the live virus vaccine. Our observations may help establish an optimized design for cultivating seed virus for live vaccine production, which depends upon the knowledge of when and why the virus evolved its virulent gene, and the overall time course of mutations during consecutive passage in a given cell line. We hope that our findings are useful in breeding a seed virus for other live vaccine production, i.e., influenza, Japanese encephalitis, etc. In conclusion, HAV is an effective model for identifying hotspots, as well as the time course and direction of evolution of the virus in given conditions. The mechanism and possible implications of this type of evolution merits further investigation. Studies utilizing other viruses should be carried out to further evaluate this type of evolution.
Footnotes
Supported partially by funds from Zhejiang Pukang Biotech. Ltd
S- Editor Zhu LH L- Editor Anand BS E- Editor Liu Y
References
- 1.Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666–1669. doi: 10.1126/science.1092002. [DOI] [PubMed] [Google Scholar]
- 2.Salemi M, Strimmer K, Hall WW, Duffy M, Delaporte E, Mboup S, Peeters M, Vandamme AM. Dating the common ancestor of SIVcpz and HIV-1 group M and the origin of HIV-1 subtypes using a new method to uncover clock-like molecular evolution. FASEB J. 2001;15:276–278. doi: 10.1096/fj.00-0449fje. [DOI] [PubMed] [Google Scholar]
- 3.Mao JS, Dong DX, Zhang HY, Chen NL, Zhang XY, Huang HY, Xie RY, Zhou TJ, Wan ZJ, Wang YZ. Primary study of attenuated live hepatitis A vaccine (H2 strain) in humans. J Infect Dis. 1989;159:621–624. doi: 10.1093/infdis/159.4.621. [DOI] [PubMed] [Google Scholar]
- 4.Guo R, Cao YY, Dai ZZ, Qu SR, Zhuang JY. Characteristics of a human diploid cell line, KMB-17. Zhongguo YiXue KeXueYuan XueBao. 1981;3:226–230. [PubMed] [Google Scholar]
- 5.Mao JS, Yu PH, Ding ZS, Chen NL, Huang BZ, Xie RY, Chai SA. Patterns of shedding of hepatitis A virus antigen in feces and of antibody responses in patients with naturally acquired type A hepatitis. J Infect Dis. 1980;142:654–659. doi: 10.1093/infdis/142.5.654. [DOI] [PubMed] [Google Scholar]
- 6.Siegl G, Frösner GG. Characterization and classification of virus particles associated with hepatitis A. I. Size, density, and sedimentation. J Virol. 1978;26:40–47. doi: 10.1128/jvi.26.1.40-47.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robertson BH, Jansen RW, Khanna B, Totsuka A, Nainan OV, Siegl G, Widell A, Margolis HS, Isomura S, Ito K. Genetic relatedness of hepatitis A virus strains recovered from different geographical regions. J Gen Virol. 1992;73(Pt6):1365–1377. doi: 10.1099/0022-1317-73-6-1365. [DOI] [PubMed] [Google Scholar]
- 8.Costa-Mattioli M, Cristina J, Romero H, Perez-Bercof R, Casane D, Colina R, Garcia L, Vega I, Glikman G, Romanowsky V, et al. Molecular evolution of hepatitis A virus: a new classification based on the complete VP1 protein. J Virol. 2002;76:9516–9525. doi: 10.1128/JVI.76.18.9516-9525.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Najarian R, Caput D, Gee W, Potter SJ, Renard A, Merryweather J, Van Nest G, Dina D. Primary structure and gene organization of human hepatitis A virus. Proc Natl Acad Sci USA. 1985;82:2627–2631. doi: 10.1073/pnas.82.9.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graff J, Normann A, Feinstone SM, Flehmig B. Nucleotide sequence of wild-type hepatitis A virus GBM in comparison with two cell culture-adapted variants. J Virol. 1994;68:548–554. doi: 10.1128/jvi.68.1.548-554.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen JI, Ticehurst JR, Purcell RH, Buckler-White A, Baroudy BM. Complete nucleotide sequence of wild-type hepatitis A virus: comparison with different strains of hepatitis A virus and other picornaviruses. J Virol. 1987;61:50–59. doi: 10.1128/jvi.61.1.50-59.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen JI, Rosenblum B, Ticehurst JR, Daemer RJ, Feinstone SM, Purcell RH. Complete nucleotide sequence of an attenuated hepatitis A virus: comparison with wild-type virus. Proc Natl Acad Sci USA. 1987;84:2497–2501. doi: 10.1073/pnas.84.8.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemon SM, Murphy PC, Shields PA, Ping LH, Feinstone SM, Cromeans T, Jansen RW. Antigenic and genetic variation in cytopathic hepatitis A virus variants arising during persistent infection: evidence for genetic recombination. J Virol. 1991;65:2056–2065. doi: 10.1128/jvi.65.4.2056-2065.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang CL, Wang PF, Liu JY, Zhang HY, Wan ZJ. Genomic sequence of hepatitis A virus L-A-1 vaccine strain. Zhonghua ShiYan HeLin ChuangBing DuXue ZaZhi. 2004;18:360–362. [PubMed] [Google Scholar]
- 15.Paul AV, Tada H, von der Helm K, Wissel T, Kiehn R, Wimmer E, Deinhardt F. The entire nucleotide sequence of the genome of human hepatitis A virus (isolate MBB) Virus Res. 1987;8:153–171. doi: 10.1016/0168-1702(87)90026-8. [DOI] [PubMed] [Google Scholar]
- 16.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Retief JD. Phylogenetic analysis using PHYLIP. Methods Mol Biol. 2000;132:243–258. doi: 10.1385/1-59259-192-2:243. [DOI] [PubMed] [Google Scholar]
- 18.Kumar S, Tamura K, Nei M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- 19.Graff J, Kasang C, Normann A, Pfisterer-Hunt M, Feinstone SM, Flehmig B. Mutational events in consecutive passages of hepatitis A virus strain GBM during cell culture adaptation. Virology. 1994;204:60–68. doi: 10.1006/viro.1994.1510. [DOI] [PubMed] [Google Scholar]
- 20.Emerson SU, Huang YK, Nguyen H, Brockington A, Govindarajan S, St Claire M, Shapiro M, Purcell RH. Identification of VP1/2A and 2C as virulence genes of hepatitis A virus and demonstration of genetic instability of 2C. J Virol. 2002;76:8551–8559. doi: 10.1128/JVI.76.17.8551-8559.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown EA, Day SP, Jansen RW, Lemon SM. The 5' nontranslated region of hepatitis A virus RNA: secondary structure and elements required for translation in vitro. J Virol. 1991;65:5828–5838. doi: 10.1128/jvi.65.11.5828-5838.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown EA, Day SP, Jansen RW, Lemon SM. Genetic variability within the 5' nontranslated region of hepatitis A virus RNA. Implications for secondary structure and function. J Hepatol. 1991;13 Suppl 4:S138–S143. doi: 10.1016/0168-8278(91)90046-e. [DOI] [PubMed] [Google Scholar]
- 23.Graff J, Emerson SU. Importance of amino acid 216 in nonstructural protein 2B for replication of hepatitis A virus in cell culture and in vivo. J Med Virol. 2003;71:7–17. doi: 10.1002/jmv.10457. [DOI] [PubMed] [Google Scholar]
- 24.Jansen RW, Newbold JE, Lemon SM. Complete nucleotide sequence of a cell culture-adapted variant of hepatitis A virus: comparison with wild-type virus with restricted capacity for in vitro replication. Virology. 1988;163:299–307. doi: 10.1016/0042-6822(88)90270-x. [DOI] [PubMed] [Google Scholar]
- 25.Morace G, Pisani G, Beneduce F, Divizia M, Panà A. Mutations in the 3A genomic region of two cytopathic strains of hepatitis A virus isolated in Italy. Virus Res. 1993;28:187–194. doi: 10.1016/0168-1702(93)90135-a. [DOI] [PubMed] [Google Scholar]
- 26.Cohen JI, Rosenblum B, Feinstone SM, Ticehurst J, Purcell RH. Attenuation and cell culture adaptation of hepatitis A virus (HAV): a genetic analysis with HAV cDNA. J Virol. 1989;63:5364–5370. doi: 10.1128/jvi.63.12.5364-5370.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raychaudhuri G, Govindarajan S, Shapiro M, Purcell RH, Emerson SU. Utilization of chimeras between human (HM-175) and simian (AGM-27) strains of hepatitis A virus to study the molecular basis of virulence. J Virol. 1998;72:7467–7475. doi: 10.1128/jvi.72.9.7467-7475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crowe JE, Bui PT, Siber GR, Elkins WR, Chanock RM, Murphy BR. Cold-passaged, temperature-sensitive mutants of human respiratory syncytial virus (RSV) are highly attenuated, immunogenic, and protective in seronegative chimpanzees, even when RSV antibodies are infused shortly before immunization. Vaccine. 1995;13:847–855. doi: 10.1016/0264-410x(94)00074-w. [DOI] [PubMed] [Google Scholar]
- 29.Konduru K, Kaplan GG. Stable growth of wild-type hepatitis A virus in cell culture. J Virol. 2006;80:1352–1360. doi: 10.1128/JVI.80.3.1352-1360.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]