

Abstract
AIM: Gastrointestinal dysfunction is a common complication in patients with traumatic brain injury (TBI). However, the effect of traumatic brain injury on intestinal mucosa has not been studied previously. The aim of the current study was to explore the alterations of intestinal mucosa morphology and barrier function, and to determine how rapidly the impairment of gut barrier function occurs and how long it persists following traumatic brain injury.
METHODS: Male Wistar rats were randomly divided into six groups (6 rats each group) including controls without brain injury and traumatic brain injury groups at hours 3, 12, 24, and 72, and on day 7. The intestinal mucosa structure was detected by histopathological examination and electron microscopy. Gut barrier dysfunction was evaluated by detecting serum endotoxin and intestinal permeability. The level of serum endotoxin and intestinal permeability was measured by using chromogenic limulus amebocyte lysate and lactulose/mannitol (L/M) ratio, respectively.
RESULTS: After traumatic brain injury, the histopathological alterations of gut mucosa occurred rapidly as early as 3 hours and progressed to a serious state, including shedding of epithelial cells, fracture of villi, focal ulcer, fusion of adjacent villi, dilation of central chyle duct, mucosal atrophy, and vascular dilation, congestion and edema in the villous interstitium and lamina propria. Apoptosis of epithelial cells, fracture and sparseness of microvilli, loss of tight junction between enterocytes, damage of mitochondria and endoplasm, were found by electron microscopy. The villous height, crypt depth and surface area in jejunum decreased progressively with the time of brain injury. As compared with that of control group (183.7 ± 41.8 EU/L), serum endotoxin level was significantly increased at 3, 12, and 24 hours following TBI (434.8 ± 54.9 EU/L, 324.2 ± 61.7 EU/L and 303.3 ± 60.2 EU/L, respectively), and peaked at 72 hours (560.5 ± 76.2 EU/L), then declined on day 7 (306.7 ± 62.4 EU/L, P < 0.01). Two peaks of serum endotoxin level were found at hours 3 and 72 following TBI. L/M ratio was also significantly higher in TBI groups than that in control group (control, 0.0172 ± 0.0009; 12 h, 0.0303 ± 0.0013; 24 h, 0.0354 ± 0.0025; 72 h, 0.0736 ± 0.0105; 7 d, 0.0588 ± 0.0083; P < 0.01).
CONCLUSION: Traumatic brain injury can induce significant damages of gut structure and impairment of barrier function which occur rapidly as early as 3 hours following brain injury and lasts for more than 7 days with marked mucosal atrophy.
INTRODUCTION
Gastrointestinal dysfunction occurs frequently in patients with traumatic brain injury (TBI)[1]. Dysfunction of the different segments of gastrointestinal tract leads to corresponding symptoms such as gastrointestinal bleeding[2-4], gastric reflux[5,6] and decreased intestinal peristalsis[7,8], mainly due to mucosal damage and alteration of gastrointestinal motility[9]. Studies on gastrointestinal dysfunction associated with head injury in the literature concentrated on the gastric mucosa, gastric emptying and lower esophageal sphincter tone[2-8]. However, the effect of TBI on intestinal mucosa has not been studied to date. Therefore, the alterations of intestinal mucosa morphology and barrier function following TBI remain unclear.
It is generally accepted that the intestine may serve as an important organ in the development of severe complications under critically ill conditions, including trauma, burns, shock, etc.[10-12]. Major trauma and shock may initiate a cascade of intestinal events such as intestinal cytokine response[13], increased intestinal permeability[14,15], translocation of intestinal bacteria and endotoxin[10,16], leading to systemic inflammatory response syndrome (SIRS) and sepsis with subsequent multiple organ failure[12]. TBI is a severe pathological stress and critically ill condition usually complicated with gastric stress ulcer[17]. Therefore, we hypothesized that TBI could also lead to significant gut structural alterations and barrier dysfunction as mentioned above in the same mechanism as trauma, hemorrhagic shock and burns. Additionally, brain-gut axis[18,19] and hypothalamic-pituitary adrenal axis[20] may also play a certain role in the development of gut dysfunction following TBI, but the potential mechanism underlying this action is not clear.
Studies have shown that cerebral inflammation following TBI occurs frequently and is associated with adverse outcomes[21,22]. Bacterial endotoxin (lipopolysaccharide, LPS) may exacerbate the inflammatory response of injured brain, leading to invasion of granulocytes into the brain and breakdown of the blood-brain barrier[23-26]. Severe extra-cerebral trauma can induce the expression of TNF-α mRNA in the brain of mice[27]. Therefore, we suggest that intestinal cytokine, translocation of bacteria and endotoxin secondary to gut barrier dysfunction into the injured brain through blood circulation should activate the cerebral inflammatory response. Because enteral nutrition and enterogenous sepsis are two of the important factors affecting the outcomes of comatose patients with TBI, the maintenance of normal gut integrity is believed to be beneficial in improving the outcomes of head-injured patients[28,29].
Considering the important role of intestinal function in the rehabilitation of patients with TBI and no study reported previously on the intestinal aspects following TBI, additional studies need to be carried out to clarify the degree of mucosal damage and gut barrier dysfunction induced by TBI, and if possible, to explore the potential mechanisms that have not been interpreted yet. Therefore, we performed a series of studies on intestines of TBI rats. The purpose of this study was to explore the alterations of intestinal mucosa morphology and barrier function, and to determine how rapidly impairment of gut barrier function occurred and how long it persisted following TBI.
MATERIALS AND METHODS
Rat models of TBI
Male Wistar rats, weighing 220 g to 250 g, were purchased from Animal Center of Chinese Academy of Sciences, Shanghai, China. The rats were fed on rodent chow with free access to tap water and housed in temperature- and humidity-controlled animal quarters with a 12 hour light/dark cycle. All procedures were approved by the Institutional Animal Care Committee.
The rats were randomly divided into six groups (6 rats each group) including control group with right parietal bone window alone and no brain injury, and traumatic brain injury groups at hours 3, 12, 24, and 72, and on day 7, respectively. Following intraperitoneal anesthesia with urethane (1000 mg.kg- 1), animal head was fixed in the stereotactic device. A right parietal bone window of 5 mm diameter was made under aseptic conditions with dental drill just behind the cranial coronal suture and beside midline. The dura was kept intact. Right parietal brain contusion was adopted using the impact method described by Feeney et al[30] and severe traumatic brain injury was made by dropping weight with a 4 mm impact diameter, a 5 mm depth and total impact energy of 1000 g.cm. The control animals were killed for sample collection at 72 hours, and TBI group rats were decapitated at corresponding time points. Blood samples were obtained by right ventricle puncture and centrifuged with the plasma stored at -40 °C before the animals were sacrificed at each time point. A 3 cm segment of the jejunum 12 cm distal to Treitz ligament was taken for histopathological studies and 1 mm3 mucosa for ultrastructural observations.
Histopathological and ultrastructural examination
The 10% buffered formalin-fixed jejunum was embedded in paraffin, sectioned at 4 μm thickness with a microtome and stained with hematoxylin and eosin. The sections were examined under light microscope. Villous height, diameter of middle villous segment and crypt depth in all tissues were determined using the HPIAS-1000 colorful image analysis system (Champion Image Engineering Company, Wuhan, China). Villous surface area was calculated according to the following formula: surface area = πdh, (d: diameter, h: villous height). At least 10 well-oriented crypt-villous units per small intestinal sample were measured and averaged by a pathologist who was blind to animal groups.
Samples for electron microscopy were fixed in phosphate-buffered glutaradehyde (2.5%) and osmium tetroxide (1%). Dehydration of the mucosa was accomplished in acetone solutions at increasing concentrations. The tissue was embedded in an epoxy resin. Semithin (1 μm) sections through the mucosa were then made and stained with toluidine blue. Then 600 angstrom-thin sections were made from a selected area of tissue defined by the semithin section, and these sections were stained with lead citrate and uranyl acetate. Ultrastructures of mucosa were observed under a transmission electron microscope (JEM-1200X).
Plasma endotoxin determination
The endotoxin content in plasma samples was assayed by the chromogenic limulus amebocyte lysate (LAL) test with a kinetic modification according to the test kit procedure.
Intestinal permeability
Intestinal permeability was quantified using the lactulose/ mannitol (L/M) test as previously described[19]. Six hours before the animals were sacrificed at each time point, the rats with their bladders emptied were given the test solution of 2.5 ml by gastric tube feeding, containing 60 mg lactulose and 30 mg mannitol. All urine was collected for 5 hours through the metabolic cage and stored at -40 °C for further analysis. Urinary lactulose and mannitol were measured using high-performance liquid chromatography (Waters Co., USA). Results were expressed as a ratio of percentage of the administered dose of the two molecules excreted.
Statistical analysis
Software SPSS 11.0 was used in the statistical analysis. Each parameter was expressed as mean ± SD, and compared using one-way ANOVA analysis of variance, followed by Tukey test. The level of significance was P < 0.05.
RESULTS
Histopathology
Histopathological examination showed that the morphology of jejunal mucosa was approximately normal in control rats. After TBI, mucosal damage occurred as early as 3 hours and was maximal at 72 hours, then progressed to mucosal atrophy on day 7. The type of jejunal mucosa damages induced by TBI is shown in Table 1. Representative histopathologic sections of jejunal mucosa are shown in Figure 1, Figure 2, Figure 3 and Figure 4.
Table 1.
Main histopathological changes of jejunal mucosa after traumatic brain injury
| Type of mucosal damages | Group of TBI |
| Shedding of epithelial cells from the top of villi | 3 and 12 hours |
| Epithelial cell necrosis and disarrangement of villi | 24 and 72 hours |
| Focal mucosa ulcer with exposure of submucosal interstitium | 72 hours |
| Fusion of adjacent villi into piece | 72 hours and 7 days |
| Dilation of central chyle duct | 72 hours and 7 days |
| Inflammatory cell infiltration in intestinal wall | 72 hours and 7 days |
| Mucosal atrophy | 7 days |
| Vascular dilation, congestion and edema in villous interstitium and lamina propria | 3 hours throughout 7 days |
Figure 1.
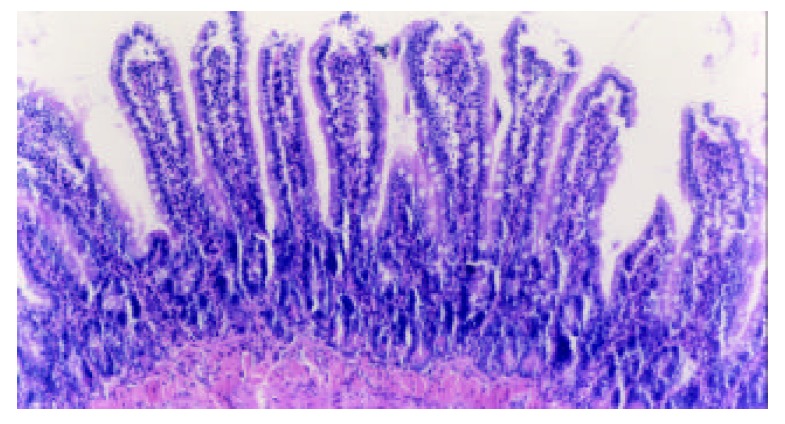
Epithelial cells shed from the top of villi with almost normal villous height and well defined arrangement of villi at 3 hours following TBI. H-E, magnification ×100.
Figure 2.
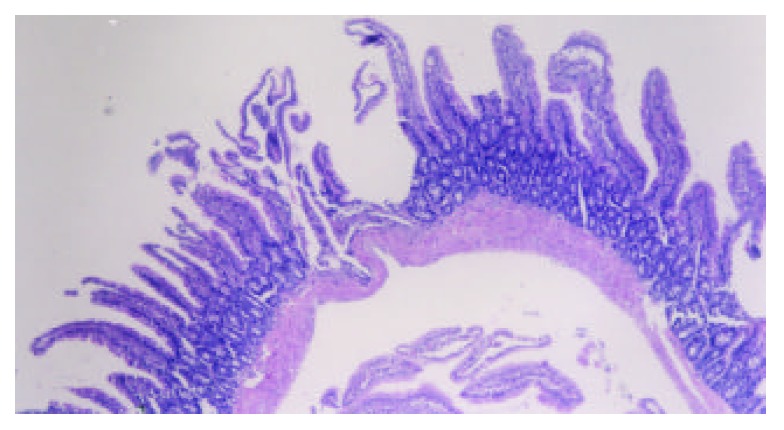
Markedly altered villous morphology and decreased height at 72 hours following TBI. Note the focal mucosa ulcer with exposure of submucosal interstitium and disarrangement of villi. H-E, magnification ×100.
Figure 3.
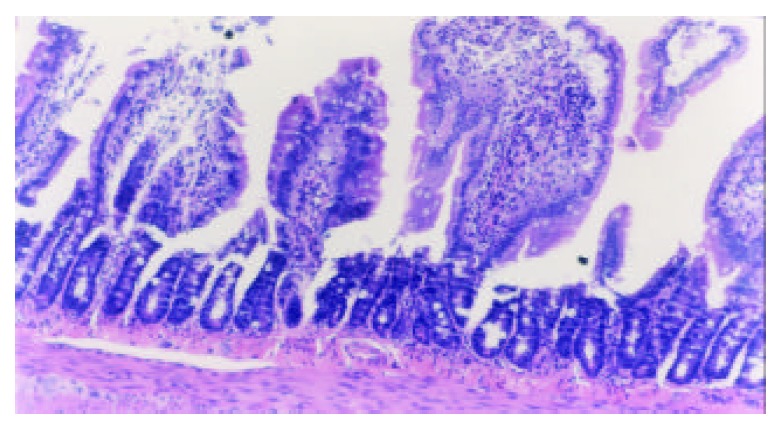
Marked alterations of villous morphology occurred 7 days following TBI, including mucosal atrophy, fusion of adjacent villi, inflammatory cell infiltration, and vascular dilation, congestion and edema in the villous interstitium and lamina propria. H-E, magnification ×100.
Figure 4.
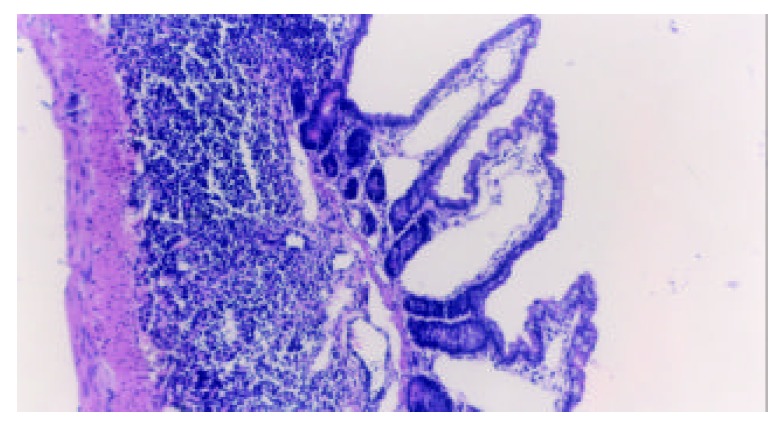
Dilation of central chyle duct of jejunal mucosa. H-E, magnification ×100.
Histomorphometric studies
Villous height, crypt depth and villous surface area were determined as specific indices for the evaluation of mucosal damages. The alterations of these parameters following TBI are shown in Table 2. Qualitative analyses of the villi demonstrated that the villous height, crypt depth and surface area were significantly decreased at 24 hours after TBI as compared with control group and further declined to the degree of mucosal atrophy on day 7 following TBI.
Table 2.
Changes in villous height, diameter, crypt depth and surface area of mucosa
| Groups | Villous height (µm) | Villous diameter (µm) | Crypt depth (µm) | Surface area (mm2) |
| Control | 229.2 ± 18.1 | 37.4 ± 5.5 | 79.3 ± 9.6 | 0.0259 ± 0.0048 |
| TBI 3 hours | 219.5 ± 21.2 | 37.6 ± 6.1 | 78.9 ± 9.4 | 0.0258 ± 0.0035 |
| TBI 12 hours | 214.5 ± 22.5 | 35.6 ± 3.1 | 74.0 ± 6.9 | 0.0241 ± 0.0037 |
| TBI 24 hours | 189.2 ± 13.6b | 32.7 ± 3.2 | 67.0 ± 7.6bc | 0.0195 ± 0.0012bc |
| TBI 72 hours | 150.8 ± 10.4bc | 32.1 ± 4.2 | 34.8 ± 5.6bc | 0.0151 ± 0.0010bc |
| TBI 7 days | 136.7 ± 15.7bc | 30.8 ± 5.8 | 38.3 ± 6.1bc | 0.0130 ± 0.0015bc |
Note: Values were expressed as mean ± SD.
P < 0.01 vs control,
P < 0.01 vs group of 3 hour TBI.
Ultrastructural observations
Within 24 hours of TBI, the nuclei of epithelial cells appeared irregular, but their membranes were still intact. At 72 hours and on day 7 following TBI, obvious ultrastructural alterations were found, including reduction of matrix of mitochondria and disruption of their cristae (Figure 5), ruptured, distorted and sparse microvilli (Figure 6), apoptosis bodies in cytoplasms (Figure 7), disrupted and wider tight junction between epithelial cells (Figure 8), swollen and degenerated vascular endothelial cells.
Figure 5.
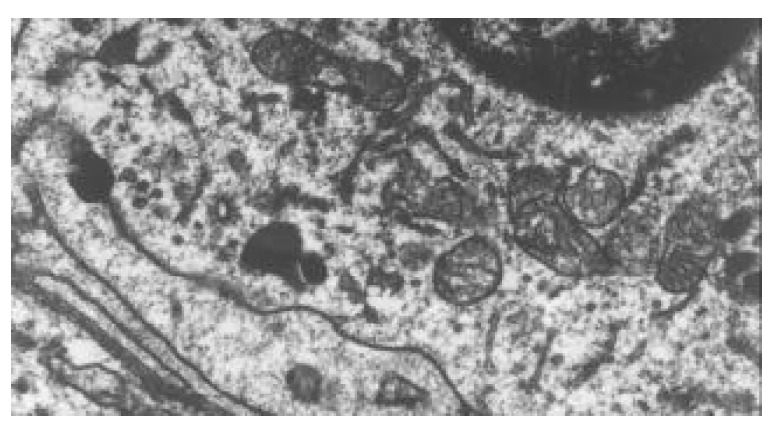
Reduction of mitochondrial matrix and disruption of its cristae at 72 hours following TBI. TEM, magnification ×15 k.
Figure 6.
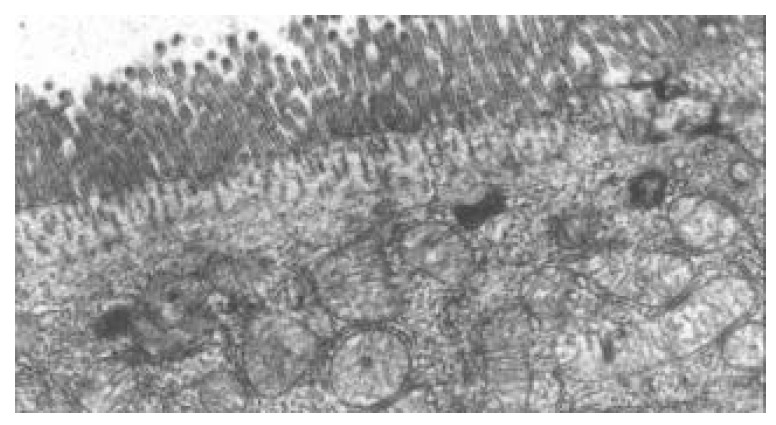
Ruptured, distorted and sparse microvilli at 24 hours following TBI. TEM, magnification ×10 k.
Figure 7.
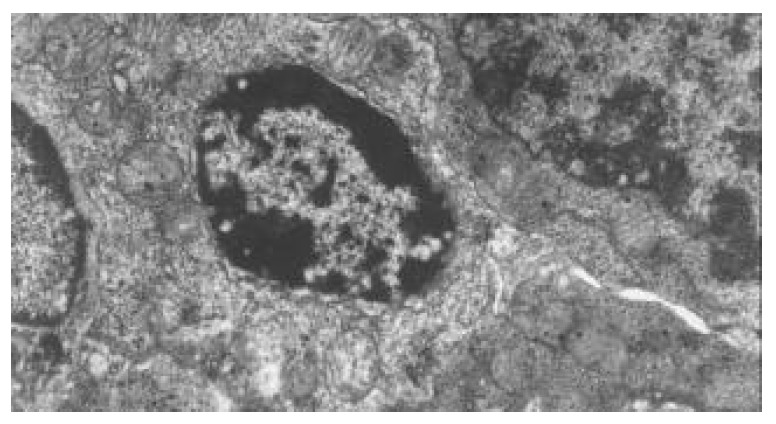
Apoptosis bodies in epithelial cytoplasms of jejunum following TBI. TEM, magnification ×8000.
Figure 8.
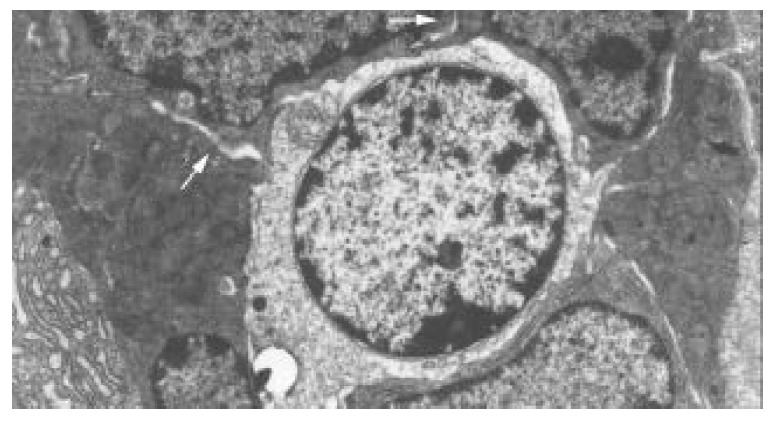
Disrupted and wider tight junction between epithe-lial cells at 72 hours and 7 days following TBI, shown as blank arrow. TEM, magnification ×5000.
Changes in plasma endotoxin concentration
As shown in Figure 9, plasma endotoxin was significantly increased at 3 hours following TBI, and peaked at 72 hours, then declined on day 7, but was still significantly higher than that of controls (P < 0.01). A marked correlation was noted between the changes in plasma level of endotoxin and intestinal permeability throughout 12 hours and 7 days following TBI (r = 0.765, P < 0.01).
Figure 9.
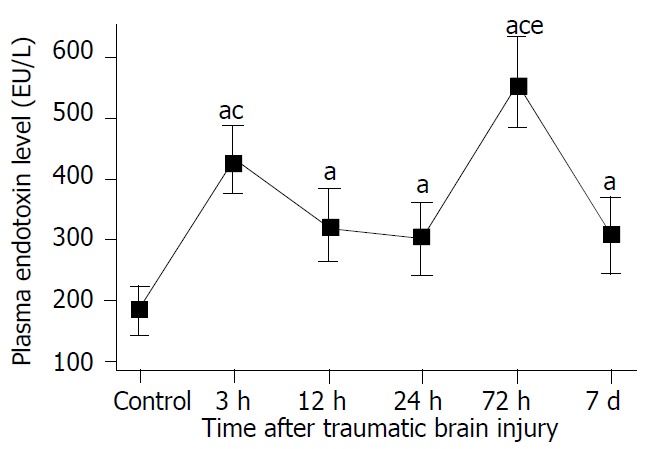
Significant increase of level of plasma endotoxin following TBI compared with control. Two peaks of plasma endotoxin existed at 3 h and 72 h postinjury, respectively. aP < 0.05 vs control; cP < 0.05 vs 12 h, 24 h and 72 h following TBI, eP < 0.05 vs 3 h following TBI. Mean ± SD of six animals, control: 183.7 ± 41.8 EU/L, 3 h: 434.8 ± 54.9 EU/L, 12 h: 324.2 ± 61.7 EU/L, 24 h: 303.3 ± 60.2 EU/L, 72 h: 560.5 ± 76.2 EU/L, 7 d: 306.7 ± 62.4 EU/L.
Intestinal permeability and L/M ratio
As shown in Figure 10, intestinal permeability was significantly increased at 12 hours after TBI, peaked at 72 hours, and declined gradually afterward, but was still higher on day 7 following TBI than that of control group.
Figure 10.
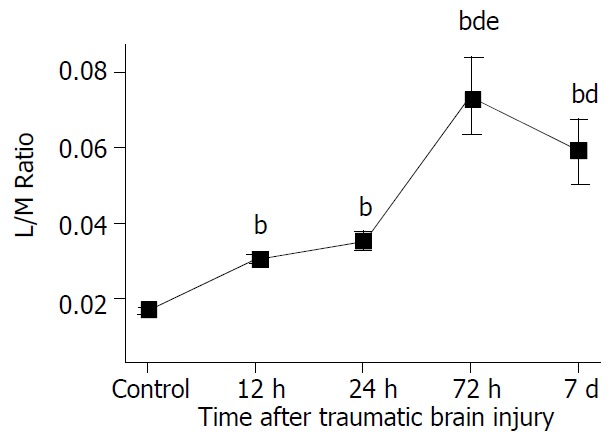
Significant increase of L/M ratio at 12 h, 24 h and 72 h following TBI compared with control and significant decline on day 7 compared with group at 72 h. The L/M ratio on day 7 was still significantly higher than that at 12 h and 24 h. bP < 0.01 vs control, dP < 0.01 vs 12 h and 24 h, eP < 0.01 vs 7 d. Mean ± SD of six animals, control: 0.0172 ± 0.0009, 12 h: 0.0303 ± 0.0013, 24 h: 0.0354 ± 0.0025,72 h: 0.0736 ± 0.0105, 7 d: 0.0588 ± 0.0083.
DISCUSSION
Major changes of gastrointestinal function following traumatic brain injury could be summarized in four aspects. The first was gastrointestinal mucosa ischemia, which usually resulted in stress ulcer and gastrointestinal bleeding[3,4]. The second was motility dysfunction, which manifested as abdominal distention, diarrhea, gastric retention, and even toxic intestinal paralysis[5-7,31]. The third was disruption of gut barrier, which led to bacterial and endotoxin translocation contributing to the development of SIRS and sepsis. The fourth was alteration of intestinal mucosal absorption of nutrients, which played an important role in malnutrition. Studies on stress ulcer and gastrointestinal bleeding after brain injury have been reported extensively[2]. Stress ulcer was one of the most common gastrointestinal complications following head trauma with a reported incidence of 74%-100%[3,4]. Endoscopic evidences of gastric mucosal lesions could appear within 24 hours, and 17% of these erosions would progress to clinically significant gastrointestinal bleeding after severe brain injury.
Studies on intestinal mucosa structure and barrier function following TBI have not been found to date. In this study, we found that severe damage of intestinal mucosa occurred following TBI. The morphologic alterations of gut mucosa included shedding of epithelial cells, fracture of villi, focal ulcer, fusion of adjacent villi, dilation of central chyle duct, mucosal atrophy, and edema in the villous interstitium and lamina propria. Apoptosis of epithelial cells, fracture and sparseness of microvilli, disruption of tight junction between enterocytes, damage of mitochondria and endoplasm, were found by electron microscopy. These lesions occurred rapidly as early as 3 hours following TBI and lasted for more than 7 days. At the early stage (within 72 hours following TBI), it manifested as acute damages of intestinal epithelium, such as epithelial necrosis and focal mucosa ulcer. On day 7 after TBI, the intestinal lesions were dominated by mucosal atrophy and edema of villous interstitium and lamina propria. Inflammatory cell infiltration in the small intestine was found 24 hours after TBI. Mucosal atrophy was considered to be associated with the necrosis and apoptosis of gut epithelium, which were induced by relative hypoperfusion (ischemia-reperfusion injury) and interaction of inflammatory mediators with their receptors located on the gut epithelial cells[13].
Intestinal mucosal injury could also be assessed by measuring the permeability of mucosal barrier to small or large solutes. Lactulose and mannitol have previously been used to assess intestinal mucosal permeability in burn and critically ill patients[12,14,15]. Mannitol, a smaller sugar, passes through aqueous pores in the cell membranes. Lactulose, a larger molecule, is absorbed paracelluarly through tight junctions. Increased absorption of lactulose can reflect mucosal leakiness, and decreased absorption of mannitol can reflect decreased functional absorptive area. The current study demonstrated that the L/M ratio was significantly greater at 3 hours following TBI, and reached its peak at 72 hours, then declined on day 7 with the value still markedly higher than that of control group and 24 hour TBI group. Additionally, the results showed that increased intestinal permeability might persist for 7 days in rats with TBI, which conformed highly to the histopathological alterations, i.e. mucosal atrophy and disruption of tight junction between epithelial cells.
The level of plasma endotoxin was positively related to L/M ratio, and manifested as two peaks at 3 hours and 72 hours after TBI, respectively. The first peak might be the result of acute gut mucosal damage mainly induced by splanchic ischemia due to the excited sympathetic nerve. With the resumption of liver anti-toxic function and the advent of specific antibodies to lipopolysaccharide, the plasma level of endotoxin was declined to some extent[34]. The second peak of plasma endotoxin might be related to severe mucosal damages such as focal ulcer and epithelial necrosis which usually occurred at 72 hours following TBI. High L/M ratio and plasma level of endotoxin following TBI implied that the gut barrier function was disrupted.
The potential mechanism underlying the alterations of gut structure and impairment of barrier function following TBI might be related to several factors, including ischemia-reperfusion, intramucosal acidosis[32], cytokines and inflammatory mediators[13,33]. Splanchnic hypoperfusion is a common finding in trauma. In systemic pathological stresses such as TBI, an adaptive response mediated by neuro-endocrine exists, which leads to selective splanchic vascular spasm in order to maintain the normal supply of vital organs such as the heart, lung and brain. The gut is highly susceptible to the consequent reduction in oxygenation, particularly mucosa of the stomach and intestine. This might induce increased gut permeability, alteration of enteral immune function, mucosal edema, epithelial necrosis and apoptosis, and even focal mucosa ulcer, which would contribute to bacterial and endotoxin translocation[13,14,20]. Brain-gut axis and hypothalamus-pituitary-adrenal axis might play a potential role in gut mucosal damages[18-20]. It may arise from the actions of parasympathetic centers of the hypothalamus with their connections to vagal nuclei in the medulla. Cytokines and inflammatory mediators such as TNF-α, IL-1, oxygen free radicals and nitric oxide could induce damages of microvilli, tight junction between enterocytes and paracelluar junction, which would lead to increased intestinal permeability[33].
Increased intestinal permeability has been implicated in the pathogenesis of both SIRS and progression to MODS[10,11,14,15]. The gut origin hypothesis suggests that a failure of gut barrier function as a result of a major stress insult permits bacterial and endotoxin translocation, which triggers systemic immunoinflammatory response to release cytokines and inflammatory mediators. These cytokines and mediators might exacerbate SIRS, sepsis and multiple organ failure[10-13]. This concept was supported by the data from animal studies in experimental models, including surgical trauma[14] , malnutrition, jaundice, pancreatitis, hemorrhagic shock, and thermal injury[28]. Results of the current study showed that gut barrier dysfunction occurred with high plasma level of endotoxin and increased intestinal permeability following TBI. Therefore, we suggest that SIRS and MODS secondary to traumatic brain injury be possibly related to the disruption of gut barrier. Otherwise, translocated endotoxin and gut-derived cytokines may enter the injured brain with circulating blood, which was assumed to activate or exacerbate the inflammation of brain, and cause “two-hit” insult[22-26]. Circulating lipopolysaccharide and cytokines may stimulate the hypothalamic-pituitary-adrenal axis and sympathetic nervous system to affect the function of multiple organs. MODS is one of the common fatal complications following severe TBI.
ACKNOWLEDGEMENTS
We would like to thank Dr. Genbao Feng, Fangnan Liu and Bo Wu for their technical assistance.
Footnotes
Supported by the Scientific Research Foundation of the Chinese PLA Key Medical Programs during the 10th Five-Year Plan Period, No. 01Z011
Edited by Zhang JZ and Wang XL
References
- 1.Pilitsis JG, Rengachary SS. Complications of head injury. Neurol Res. 2001;23:227–236. doi: 10.1179/016164101101198389. [DOI] [PubMed] [Google Scholar]
- 2.Lu WY, Rhoney DH, Boling WB, Johnson JD, Smith TC. A review of stress ulcer prophylaxis in the neurosurgical intensive care unit. Neurosurgery. 1997;41:416–25; discussion 425-6. doi: 10.1097/00006123-199708000-00017. [DOI] [PubMed] [Google Scholar]
- 3.Brown TH, Davidson PF, Larson GM. Acute gastritis occurring within 24 hours of severe head injury. Gastrointest Endosc. 1989;35:37–40. doi: 10.1016/s0016-5107(89)72683-3. [DOI] [PubMed] [Google Scholar]
- 4.Kamada T, Fusamoto H, Kawano S, Noguchi M, Hiramatsu K. Gastrointestinal bleeding following head injury: a clinical study of 433 cases. J Trauma. 1977;17:44–47. [PubMed] [Google Scholar]
- 5.Kao CH, ChangLai SP, Chieng PU, Yen TC. Gastric emptying in head-injured patients. Am J Gastroenterol. 1998;93:1108–1112. doi: 10.1111/j.1572-0241.1998.00338.x. [DOI] [PubMed] [Google Scholar]
- 6.Saxe JM, Ledgerwood AM, Lucas CE, Lucas WF. Lower esophageal sphincter dysfunction precludes safe gastric feeding after head injury. J Trauma. 1994;37:581–584; discussion 581-584;. doi: 10.1097/00005373-199410000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Pedoto MJ, O'Dell MW, Thrun M, Hollifield D. Superior mesenteric artery syndrome in traumatic brain injury: two cases. Arch Phys Med Rehabil. 1995;76:871–875. doi: 10.1016/s0003-9993(95)80555-9. [DOI] [PubMed] [Google Scholar]
- 8.Philip PA. Superior mesenteric artery syndrome: an unusual cause of intestinal obstruction in brain-injured children. Brain Inj. 1992;6:351–358. doi: 10.3109/02699059209034949. [DOI] [PubMed] [Google Scholar]
- 9.Jackson MD, Davidoff G. Gastroparesis following traumatic brain injury and response to metoclopramide therapy. Arch Phys Med Rehabil. 1989;70:553–555. [PubMed] [Google Scholar]
- 10.Swank GM, Deitch EA. Role of the gut in multiple organ failure: bacterial translocation and permeability changes. World J Surg. 1996;20:411–417. doi: 10.1007/s002689900065. [DOI] [PubMed] [Google Scholar]
- 11.Moore FA. The role of the gastrointestinal tract in postinjury multiple organ failure. Am J Surg. 1999;178:449–453. doi: 10.1016/s0002-9610(99)00231-7. [DOI] [PubMed] [Google Scholar]
- 12.Doig CJ, Sutherland LR, Sandham JD, Fick GH, Verhoef M, Meddings JB. Increased intestinal permeability is associated with the development of multiple organ dysfunction syndrome in critically ill ICU patients. Am J Respir Crit Care Med. 1998;158:444–451. doi: 10.1164/ajrccm.158.2.9710092. [DOI] [PubMed] [Google Scholar]
- 13.Grotz MR, Deitch EA, Ding J, Xu D, Huang Q, Regel G. Intestinal cytokine response after gut ischemia: role of gut barrier failure. Ann Surg. 1999;229:478–486. doi: 10.1097/00000658-199904000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langkamp-Henken B, Donovan TB, Pate LM, Maull CD, Kudsk KA. Increased intestinal permeability following blunt and penetrating trauma. Crit Care Med. 1995;23:660–664. doi: 10.1097/00003246-199504000-00013. [DOI] [PubMed] [Google Scholar]
- 15.Faries PL, Simon RJ, Martella AT, Lee MJ, Machiedo GW. Intestinal permeability correlates with severity of injury in trauma patients. J Trauma. 1998;44:1031–1035; discussion 1031-1035;. doi: 10.1097/00005373-199806000-00016. [DOI] [PubMed] [Google Scholar]
- 16.Wang XD, Soltesz V, Andersson R. Cisapride prevents enteric bacterial overgrowth and translocation by improvement of intestinal motility in rats with acute liver failure. Eur Surg Res. 1996;28:402–412. doi: 10.1159/000129484. [DOI] [PubMed] [Google Scholar]
- 17.Pepe JL, Barba CA. The metabolic response to acute traumatic brain injury and implications for nutritional support. J Head Trauma Rehabil. 1999;14:462–474. doi: 10.1097/00001199-199910000-00007. [DOI] [PubMed] [Google Scholar]
- 18.Glavin GB, Hall AM. Brain-gut relationships: gastric mucosal defense is also important. Acta Physiol Hung. 1992;80:107–115. [PubMed] [Google Scholar]
- 19.Shanahan F. Brain-gut axis and mucosal immunity: a perspective on mucosal psychoneuroimmunology. Semin Gastrointest Dis. 1999;10:8–13. [PubMed] [Google Scholar]
- 20.Grundy PL, Harbuz MS, Jessop DS, Lightman SL, Sharples PM. The hypothalamo-pituitary-adrenal axis response to experimental traumatic brain injury. J Neurotrauma. 2001;18:1373–1381. doi: 10.1089/08977150152725669. [DOI] [PubMed] [Google Scholar]
- 21.Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16:165–177. doi: 10.1097/00024382-200116030-00001. [DOI] [PubMed] [Google Scholar]
- 22.Ghirnikar RS, Lee YL, Eng LF. Inflammation in traumatic brain injury: role of cytokines and chemokines. Neurochem Res. 1998;23:329–340. doi: 10.1023/a:1022453332560. [DOI] [PubMed] [Google Scholar]
- 23.Bohatschek M, Werner A, Raivich G. Systemic LPS injection leads to granulocyte influx into normal and injured brain: effects of ICAM-1 deficiency. Exp Neurol. 2001;172:137–152. doi: 10.1006/exnr.2001.7764. [DOI] [PubMed] [Google Scholar]
- 24.Ahmed SH, He YY, Nassief A, Xu J, Xu XM, Hsu CY, Faraci FM. Effects of lipopolysaccharide priming on acute ischemic brain injury. Stroke. 2000;31:193–199. doi: 10.1161/01.str.31.1.193. [DOI] [PubMed] [Google Scholar]
- 25.Montero-Menei CN, Sindji L, Garcion E, Mege M, Couez D, Gamelin E, Darcy F. Early events of the inflammatory reaction induced in rat brain by lipopolysaccharide intracerebral injection: relative contribution of peripheral monocytes and activated microglia. Brain Res. 1996;724:55–66. doi: 10.1016/0006-8993(96)00268-5. [DOI] [PubMed] [Google Scholar]
- 26.Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care. 2002;8:101–105. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Kamei H, Yoshida S, Yamasaki K, Tajiri T, Ozaki K, Shirouzu K. Severity of trauma changes expression of TNF-alpha mRNA in the brain of mice. J Surg Res. 2000;89:20–25. doi: 10.1006/jsre.1999.5802. [DOI] [PubMed] [Google Scholar]
- 28.Zhu L, Yang ZC, Li A, Cheng DC. Protective effect of early enteral feeding on postburn impairment of liver function and its mechanism in rats. World J Gastroenterol. 2000;6:79–83. doi: 10.3748/wjg.v6.i1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suchner U, Senftleben U, Eckart T, Scholz MR, Beck K, Murr R, Enzenbach R, Peter K. Enteral versus parenteral nutrition: effects on gastrointestinal function and metabolism. Nutrition. 1996;12:13–22. doi: 10.1016/0899-9007(95)00016-x. [DOI] [PubMed] [Google Scholar]
- 30.Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 1981;211:67–77. doi: 10.1016/0006-8993(81)90067-6. [DOI] [PubMed] [Google Scholar]
- 31.Mackay LE, Morgan AS, Bernstein BA. Factors affecting oral feeding with severe traumatic brain injury. J Head Trauma Rehabil. 1999;14:435–447. doi: 10.1097/00001199-199910000-00004. [DOI] [PubMed] [Google Scholar]
- 32.Salzman AL, Wang H, Wollert PS, Vandermeer TJ, Compton CC, Denenberg AG, Fink MP. Endotoxin-induced ileal mucosal hyperpermeability in pigs: role of tissue acidosis. Am J Physiol. 1994;266:G633–G646. doi: 10.1152/ajpgi.1994.266.4.G633. [DOI] [PubMed] [Google Scholar]
- 33.Gong JP, Wu CX, Liu CA, Li SW, Shi YJ, Yang K, Li Y, Li XH. Intestinal damage mediated by Kupffer cells in rats with endotoxemia. World J Gastroenterol. 2002;8:923–927. doi: 10.3748/wjg.v8.i5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buttenschoen K, Berger D, Strecker W, Buttenschoen DC, Stenzel K, Pieper T, Beger HG. Association of endotoxemia and production of antibodies against endotoxins after multiple injuries. J Trauma. 2000;48:918–923. doi: 10.1097/00005373-200005000-00017. [DOI] [PubMed] [Google Scholar]