

Abstract
Histone Nuclear Factor P (HINFP) is essential for expression of histone H4 genes. Ablation of Hinfp and consequential depletion of histones alter nucleosome spacing and cause stalled replication and DNA damage that ultimately result in genomic instability. Faithful replication and packaging of newly replicated DNA are required for normal cell cycle control and proliferation. The tumor suppressor protein p53, the guardian of the genome, controls multiple cell cycle checkpoints and its loss leads to cellular transformation. Here we addressed whether the absence of p53 impacts the outcomes/consequences of Hinfp-mediated histone H4 deficiency. We examined mouse embryonic fibroblasts lacking both Hinfp and p53. Our data revealed that the reduced histone H4 expression caused by depletion of Hinfp persists when p53 is also inactivated. Loss of p53 enhanced the abnormalities in nuclear shape and size (i.e. multi-lobed irregularly shaped nuclei) caused by Hinfp depletion and also altered the sub-nuclear organization of Histone Locus Bodies (HLBs). In addition to the polyploid phenotype resulting from deletion of either p53 or Hinfp, inactivation of both p53 and Hinfp increased mitotic defects and generated chromosomal fragility and susceptibility to DNA damage. Thus, our study conclusively establishes that simultaneous loss of both Hinfp and the p53 checkpoint is detrimental to normal cell growth and may predispose to cellular transformation.
Keywords: Chromosome Fragmentation, Cell Cycle, Hinfp, histone H4, Histone Locus Bodies, NPAT, p53
Abbreviations
- HINFP
Histone Nuclear Factor P
- NPAT
Nuclear Protein Ataxia-Telangiectasia locus
- HLBs
Histone Locus Bodies
- cKO
conditional knockout
- dKO
double knockout
- MEFs
mouse embryonic fibroblasts.
Introduction
Competency for DNA replication and fidelity of cell cycle checkpoints at the G1/S or G2/M transitions are required for normal growth and proliferation potential. Deregulation of cell cycle checkpoints is a principal contributor to tumor initiation. In eukaryotic cells, genomic DNA is wrapped around an octamer of core histone subunits (H2a, H2b, H3 and H4) to package DNA into nucleosomes.1-4 Histone gene expression is upregulated at the onset of S phase to accommodate packaging of newly synthesized DNA as chromatin during replication.5-7 A highly conserved Zn finger transcription factor, histone nuclear factor P (HINFP) is critical for control of histone H4 gene expression during the cell cycle.1,7,8 HINFP interacts with NPAT (Nuclear Protein Ataxia-Telangiectasia locus), a cyclin E/cyclin-dependent kinase 2 (CDK2) effector that regulates multiple histone genes.9-14 NPAT together with HINFP is an integral component of sub-nuclear domains designated histone locus bodies (HLBs). Histone gene transcription and histone mRNA processing machinery co-localize with histone gene clusters on chromosomes 6 and 1 to form HLBs.14-18 The HINFP-NPAT complex regulates expression of histone genes during the G1/S phase and entry into S phase.9-11,19-21
Our recent findings indicated that Hinfp-mediated control of histone H4 gene expression during S phase is essential for cell growth and proliferation.22-24 Complete ablation of Hinfp in mice causes early embryonic lethality between embryonic day (E) 3.5 and E6.5. In vitro cultures of Hinfp-null embryos at E3.5 exhibit abnormal growth and proliferation.22 Our studies also demonstrated that conditional removal of Hinfp in mouse embryonic fibroblasts (MEFs) resulted in reduced expression of histone H4 transcripts as well as protein and subsequently caused a spectrum of cell cycle and proliferation defects.24 Inactivation of Hinfp and consequential reduction in histone H4 expression directly impacted nucleosomal assembly and generated replicative stress both during interphase and mitosis.24 Thus, Hinfp may be a key regulatory factor that controls genomic stability.
Eukaryotic cells utilize a series of DNA surveillance mechanisms to identify and repair DNA damage. The tumor suppressor protein p53 is an integral component of these key cellular regulatory pathways.25 In normal cells, DNA damage and replicative stress activate p53 induced cell cycle arrest.26 For example, treatment of p53 null MEFs with double strand break inducers such as doxorubicin activates cell cycle arrest in G2 and also suggests a role for p53 in S-phase checkpoint.27 Additionally, gamma-irradiation causes G1 arrest in MEFs wildtype for p53, but p53 null MEFs fail to induce the cell cycle block.28 Consequently, MEFs lacking functional p53 exhibit increased proliferative and transformation properties.29,30 These properties of p53 strongly suggest that loss of p53 may contribute to the severity of the Hinfp null phenotypes.
In this study, we addressed the consequences of genomic ablation of Hinfp in p53 null cells on cell cycle and cell proliferation. We crossed mice bearing a conditional allele of Hinfp with mice mutated for p53, and generated primary cells that were co-deleted for Hinfp and p53. Analysis of these cells revealed that removal of both Hinfp and p53 aggravates the cellular defects caused by Hinfp-mediated H4 deficiency. Our findings demonstrate that simultaneous loss of Hinfp and p53 is deleterious to normal cell growth and may predispose to cellular transformation.
Results
Compromised expression of histone H4 transcript persists after loss of p53 in Hinfp null cells
Because Hinfp deficiency compromises H4 gene expression and induces genomic instability, we investigated the cellular interplay between Hinfp and p53. Loss of p53 expression is known to accelerate cell proliferation.29 Therefore, overall expression of all histone mRNA is higher in p53 null cells and dKO cells compared to wild type cells (data not shown). Because Hinfp expression is essential for H4 gene transcription, relative expression of histone H4 mRNAs is compromised in both cKO cells lacking Hinfp only and dKO cells depleted for both Hinfp and p53 (Fig. 1A). Indeed the magnitude of the consequences of Hinfp deficiency extended to altered expression of other core and linker histone (H1, H2a, H2b, H3) genes (Fig. 1B). The data revealed transient increase in the expression of all 4 other histone subtypes (i.e., H1, H2a, H2b, H3) both in cKO and dKO cells. The simultaneous decrease of histone H4 mRNA and increase in expression of other histone subtypes indicates deregulation of normal stoichiometry of histone gene expression, as the levels of histone H4 mRNA are normally tightly coupled to those of the other histone subtypes. This deregulation may reflect an imbalance in the normal auto-regulatory mechanism that control histone mRNA accumulation and that are p53 independent. Importantly, loss of p53 does not affect the Hinfp dependent histone H4 deficiency.
Figure 1.
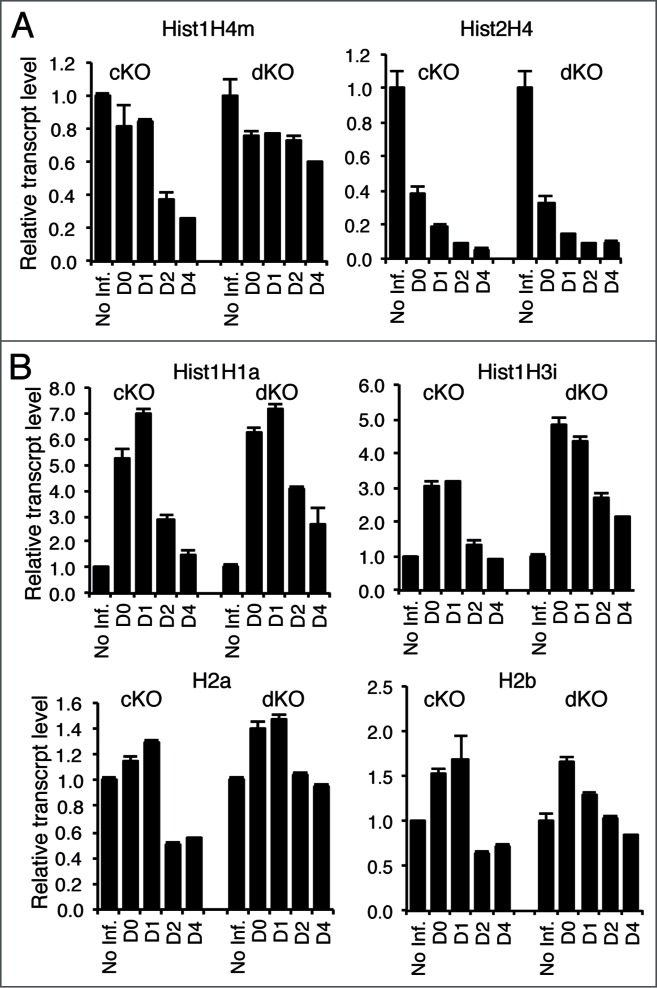
Removal of Hinfp causes reduction of histone H4 mRNA levels in both cKO and dKO cells. Quantitative PCR analysis of relative mRNA levels was performed for different histone genes in both cKO and dKO cells. (A) RNAs for 2 Hinfp-dependent histone H4 genes, Hist1H4m and Hist2H4 are reduced after Hinfp ablation, (B) Other core and linker histones upon Hinfp-mediated histone H4 depletion show transient increases in expression levels. (Hist1H1a, Hist1H3i, H2a, H2b).
Ablation of p53 in Hinfp depleted cells significantly disrupts proliferation and impairs active DNA synthesis
We have recently shown that conditional removal of histone H4 gene transcription factor Hinfp generates chromatin defects that predisposes to replicative stress and DNA damage. Because loss of Hinfp triggers a p53 dependent cell cycle checkpoint, it is important to address whether loss of p53 contributes to the phenotype of Hinfp null cells. Therefore, we examined the combined effects of loss of both Hinfp and p53 in mouse embryonic fibroblasts. While loss of Hinfp compromises cell cycle progression, loss of p53 by itself accelerates cell proliferation. Strikingly, removal of Hinfp from p53 null cells (dKO cells) causes a significant proliferation defect (p = 0.048) compared to cells that lack only p53. This dramatic change in proliferation of dKO cells is comparable to severe proliferation defect observed in cKO cells (p = 0.045) (Fig. 2A). Notably, loss of p53 in Hinfp null cells abolishes the compensatory response observed in p21 expression after ablation of only Hinfp (Fig. 2B). Thus, negative phenotypic effects of the Hinfp null mutation on cell proliferation are dominant over the stimulatory effects of p53 null mutation, which are linked to abrogation of the p53-p21 pathway.
Figure 2.

Ablation of Hinfp in p53 null cells affects cell proliferation and impairs DNA synthesis. (A) Cell growth curves show loss of Hinfp causes severe proliferation defects in both cKO (*p=0.045) and dKO (*p=0.048) cells. Wildtype (WT) cells and p53 null (−/−) cells not treated with Cre were used as comparison. (B) Q-RT-PCR showing relative mRNA levels of p21 in response to loss of Hinfp in the presence (cKO) or absence (dKO) of p53. (C) Cell cycle profiles of cKO and dKO cells exhibit progressive accumulation of polyploid cells after ablation of Hinfp. WT and p53 −/− cells are shown for comparison (left panels). (D) IF microscopy performed on WT, p53 −/−, cKO and dKO cells pulse labeled with BrdU (red). Nuclei are stained with DAPI (blue). cKO cells at D3 show a substantial decrease in BrDU incorporation, while dKO cells lacking both Hinfp and p53 show much higher frequency of BrdU incorporation. Scale Bar 100µM. (E) Quantification of BrdU incorporation. The bar graphs show percentage of cells engaged in active DNA synthesis as calculated by counting of BrdU positive cells. (F) FACS analysis of cells pulse-labeled with BrdU using BrdU-APC kit for quantitative measurements of percent S-phase in cKO and dKO cells. Note the substantial increase in the fraction of BrdU +cells after removal of both p53 and Hinfp at D2.
We further evaluated the effects of removal of both p53 and Hinfp on cell cycle properties of cKO and dKO cells by analyzing DNA content using flow cytometry. We examined the cell cycle profile of cKO and dKO cells with or without Cre treatment. The data revealed that there is a substantial population of cells with increased S-phase DNA content in both cKO and dKO cells, as observed by the increased height of the region between the G1 and G2 peaks (D1-D4). Interestingly, there is large fraction of cells with polyploid (4N/8N) DNA content in both cKO and dKO cells (Fig. 2C). While loss of p53 is known to cause a polyploid phenotype,29,31,32 the incidence of polyploidy is much higher in dKO cells lacking both Hinfp and p53. Hence, a polyploid phenotype observed in either p53 null or Hinfp null cells is further aggravated by simultaneous loss of both genes.
We next investigated active DNA synthesis in cultures of cKO and dKO cells by pulse labeling the cells with bromodeoxyuridine (BrdU). BrdU incorporation was analyzed by immunofluorescence (IF) microscopy (Fig. 2D, E) and flow cytometry (FACS) (Fig. 2F). Notably, both IF and FACS analysis revealed a higher S-phase fraction in p53 null (46.6 ± 2.3%) versus wild type (22.2 ± 2.2%) cells. This result could be explained by inherently higher proliferative properties observed in cells lacking p53. Our data also revealed that depletion of Hinfp impedes active DNA synthesis (e.g., 9.5% at D3 for cKO) as expected. However, cells lacking both Hinfp and p53 show a higher percentage of BrdU positive cells (e.g.,, 45.2% at D3 for dKO). These findings suggest that even though proliferation is significantly compromised by loss of Hinfp, concomitant loss of the p53 ablates the normal cell cycle arrest that is linked to the p53-p21 pathway resulting in promiscuous entry into S-phase.
Loss of p53 checkpoint further enhances the aberrant nuclear phenotypes observed in Hinfp depleted cells
We examined the cellular morphology of the conditional knockout and double knockout cells by immunofluorescence microscopy because forward scatter measurements suggested an increase in cell size (see Fig. 2). Nuclear staining with DAPI revealed loss of Hinfp in both cKO and dKO cells generates a much higher percentage of nuclei that are larger in size and more irregularly shaped (fused and/or multi-lobed). It appears that cells lacking Hinfp phenotypically progress to a bi-nucleate morphology and the additional loss of p53 transitions the cells into a multi-lobed nuclear morphology (Fig. 3A, B). At early stages in culture we find that dKO cells lacking both Hinfp and p53 have 50–100% more bi-nucleate cells than cKO cells (at D1: cKO = 12.5% and dKO = 24%; at D2: cKO = 24.5% and dKO = 33%). At later stages of the culture, the number of irregularly shaped nuclei in dKO cells is ∼10 times higher than cKO cells (at D4: cKO = 4.5% and dKO = 47.5%). The observed nuclear changes were also examined in the context of cellular architecture using α-tubulin staining. The images revealed that dKO cells have a higher percentage of multi-lobed nuclei compared to cKO cells (Fig. 3C). As denoted by α-tubulin staining, the typical elongated fibroblastic appearance of cKO cells was severely altered in dKO cells (Fig. 3C, lower panels). Taken together, the findings from FACS analysis and IF revealed that loss of the p53 checkpoint enhances the defects in nuclear morphology and cell proliferation caused by depletion of Hinfp alone.
Figure 3.

Simultaneous loss of p53 aggravates the aberrant nuclear morphological phenotypes of Hinfp-ablated cells. (A) IF microscopy reveals drastic changes in nuclear size and shape of cKO MEFs that are exacerbated in dKO MEFs. Nuclei stained with DAPI show increased appearance of irregularly shaped morphology in cells lacking both p53 and Hinfp. Scale Bar 50µm. (B) The bar graph shows distribution of different nuclear morphologies in cKO and dKO MEFs (n = 200 ). (C) IF microscopy of cKO and dKO MEFs stained with α-Tubulin (red) shows increased presence of bi-nucleated cells (white arrowheads) in cKO MEFs. The dKO MEFs have an enhanced phenotype with higher incidence of irregularly shaped fused nuclei (yellow arrowheads). Scale Bar 50 µm.
Removal of p53 in Hinfp null cells alters histone locus bodies (HLBs), induces DNA damage and enhances genomic instability
Because Hinfp is required for transcription of histone H4 genes and proper assembly of histone locus bodies, we evaluated the effects of depleting both Hinfp and p53 on organization of HLBs using NPAT as a marker. Depending on the cell cycle stage, normal diploid cells have 2 or 4 bright NPAT foci/HLBs per nucleus that coincide with the location of the 2 major histone gene clusters on 6p21 and 1q22. At early culture stages, the majority of cKO cells (at D1: ∼85%) have a normal HLB pattern that is progressively lost (at D4 ∼25%) due to loss of Hinfp. In contrast, dKO cells at D1 show normal HLB patterns in only ∼60% of nuclei. Simultaneously, cKO cells exhibit an increasing fraction of nuclei with diffused NPAT (at D4: 47.5%), indicating disorganized HLBs. Of note, in dKO cells lacking both p53 and Hinfp, there are few cells with diffused NPAT staining (at D4: 5.5%) (Fig. 4A, B). Interestingly, the fraction of cKO cells with more than 4 NPAT foci per nucleus (D1: 11.5%, D2: 23%, D4: 25.5%) was dramatically increased in dKO cells lacking both p53 and Hinfp (D1: 42%, D2: 65.5%, D4: 76.5%). The fraction of cells with more than 4 NPAT foci correlates with the extent of polyploidy observed by FACS analysis in cKO and dKO cells (see Fig. 3). Taken together, these results indicate that loss of Hinfp disrupts assembly of HLBs and additional loss of the p53 checkpoint deregulates HLB number per nucleus.
Figure 4.
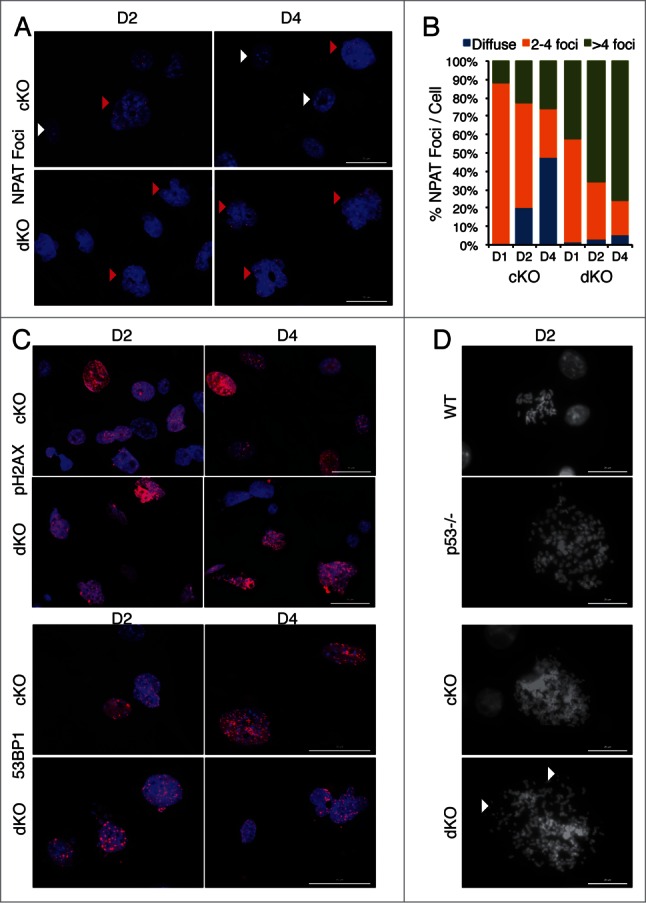
Removal of p53 in Hinfp null cells alters histone locus body (HLB) patterns and induces genomic instability. (A) IF microscopy shows the distribution of HLB by staining for the marker protein NPAT (red), an integral component of HLBs. cKO MEFs show an increase in the fraction of cells with multiple NPAT foci (red arrowheads) or diffused NPAT staining (white arrowheads) after Hinfp depletion. dKO MEFs show a further increase in cells with multiple NPAT foci. Nuclei were co-stained with DAPI (blue). Scale Bar 50 µm. (B) Quantitation of NPAT staining patterns in cKO and dKO MEFs at d1, d2 and d4 after removal of Hinfp. Nuclei were counted from 2 biological replicates (200 each) per sample for each time point. (C) cKO and dKO MEFs were analyzed for factors associated with double-strand DNA damage using immunofluorescence microscopy. (Upper panels) γ-H2Ax S-139 (red) and Scale Bar 50 µm. (Lower panels) 53BP1 (red) Scale Bar 50 µm. Nuclei were co-stained with DAPI (blue). Both cKO and dKO MEFs show cells with focal staining for both γ-H2Ax and 53BP1 that is even higher in dKO cells. (D) WT, p53−/−, cKO and dKO MEFs were mitotically arrested at D2 using Colcemid (100 ng/ml). The mitoses were analyzed by fluorescence microscopy. The DNA was stained with DAPI. cKO cells have an increased chromosome complement compared to WT, and there is increased appearance of chromosomal fragility (white arrowheads) in cells lacking both p53 and Hinfp. Scale Bar 20 µm.
We also examined the extent of DNA damage in cKO and dKO cells by investigating 2 markers closely associated with spontaneous double strand DNA breaks, γ-H2AX foci (phosphorylation of H2AX at S-139) and distribution of 53BP1 nuclear staining.33,34 Both Hinfp depleted and double knockout cells show robust, focal staining for γ-H2AX and 53BP1. However, a higher percentage of dKO cells have focal nuclear staining for both markers (Fig. 4C) indicating an enhanced level of DNA damage. Alterations in nuclear size and the probability of increased genomic instability prompted us to investigate the integrity of chromosomes in response to loss of p53 (Fig. 4D). We mitotically blocked both cKO and dKO MEFs at D2 after removal of Hinfp, as well as wildtype (WT) and p53 null cells for comparison. The morphology of mitotic chromosomes was microscopically evaluated by DAPI staining. Hinfp depleted cells show an increased chromosome complement consisting of mitoses with polyploidy (cKO: ∼40–50%) compared to wild type cells (WT: ∼10%). Embryonic fibroblasts null for p53 are known to have a higher percentage of cells with 4n chromosome complement (p53 null: ∼50%),29,31,32 which is further enhanced by loss of Hinfp (dKO: ∼90%). Interestingly, the morphology of mitotic chromosomes in cKO and dKO cells show a striking difference – Hinfp depleted cells have chromosomes with a more thread-like and loosely compacted morphology, whereas the dKO cells have fragmented chromosomes (Fig. 4D). These results suggest that loss of p53 in Hinfp depleted cells creates fragile chromosomes that are prone to breakage. Taken together, these findings indicate that loss of the p53 checkpoint worsens the chromosomal aberrations and nuclear dysmorphology caused by -Hinfp-mediated histone H4 deficiency.
Discussion
We have previously demonstrated that loss of Hinfp depletes histone H4 in chromatin and causes major defects during DNA replication that ultimately result in genomic instability.24 Because p53 is the guardian of the genome,35 our current study addressed whether p53 function is functionally linked to the phenotype of Hinfp deficient cells. Loss of Hinfp alters H4 gene transcription and stoichiometric auto-regulatory mechanisms that control histone mRNA accumulation. We find that loss of p53 does not affect this deregulation of histone H4 gene transcription observed in Hinfp null cells. However, loss of the p53-p21 pathway in Hinfp null cells bypasses a geno-protective cell cycle arrest and results in precocious entry into S-phase. Furthermore, p53 inactivation intensifies changes in nuclear morphology and sub-nuclear organization linked to Hinfp loss of function. Our findings are consistent with a mechanistic model in which p53 function mitigates Hinfp-related histone H4 deficiency and concomitant chromatin defects. Replication of H4 deficient chromatin in the absence of p53 generates chromosomal fragility and susceptibility to damage. Thus, our study conclusively establishes that simultaneous loss of both Hinfp and the p53 checkpoint is detrimental to normal cell growth and may predispose to oncogenic transformation.
Apart from deleterious defects from p53 deletion that precociously licenses replication of damaged chromatin during S-phase prior to repair, loss of p53 also has major effect on mitosis. Because p53 is the guardian of the genome and a major tumor suppressor, it coordinates several cellular checkpoints linked to G1/S transition and mitotic entry.36,37 Key findings of our study are the mechanisms by which p53 compounds the Hinfp phenotype. Loss of Hinfp generates polyploidy and reduced cell viability. However, when p53 is simultaneously ablated it exacerbates polyploidy because the p53 controlled mitotic checkpoint is compromised. We have recently shown that depletion of Hinfp causes polyploidy by triggering mitoses with multipolar spindles.24 While such aberrant divisions would normally be prevented or mitigated by p53, ablation of p53 in Hinfp null cells generates polyploid cells with fragile chromosomes. This chromosomal fragility may further account for the chromosomal imbalance and aneuploidy that we observed in cells lacking both p53 and Hinfp.
Our finding that loss of Hinfp results in chromosomal defects, suggests that wild type Hinfp may have tumor suppressor effects by maintaining chromatin integrity and ensuring the proper stoichiometry between histone gene expression and DNA replication. This protective function of Hinfp is analogous to that of p53, which is the frequently mutated in cancer (i.e., >50% of cancers exhibit p53 mutations or functional inactivation).38,39 Cells with p53 null mutation exhibit faster proliferation and higher survival potential, because p53 controls cell survival/apoptotic pathways (e.g., p53/p21 checkpoint).40,41 Unlike loss of p53, Hinfp inactivation does not confer a proliferative advantage because it is necessary for chromatin integrity and fidelity of nucleosomal organization. Consequently, there are no reports that suggest Hinfp is genetically linked to cancer. Because chromosomal defects caused by Hinfp deficiency cannot be repaired by p53 dependent checkpoints at the G1/S phase transition and mitosis, it is evident that Hinfp is an essential transcription factor required for both normal cell growth and proliferation of prevalent cancers with p53 mutations.
Materials and Methods
Mice
Conditional HinfpF/F24 and p53 knockout42 mice were crossed to generate mice both homozygous floxed for Hinfp (HinfpF/F) and p53-heterozygous. Animals were maintained according to Institutional Animal Care (IACUC) guidelines. To confirm the genotypes of the mice, tail DNA from pups was subjected to Southern blotting (data not shown) and genomic DNA PCR (Fig. S1A).
Cell culture and flow cytometry
Mouse embryonic fibroblasts (MEFs) were isolated at E13.5 from embryos with genotypes: wildtype, HinfpWTp53−/−, cHinfpF/Fp53WT, and cHinfpF/Fp53−/−. cHinfp MEFs were cultured and treated with adenoviral vector expressing Cre recombinase (Ad5CMVCre-eGFP; http://www.medicine.uiowa.edu/vectorcore/ad/stock/) at passage 3. Cre recombination of HinfpF/F produced Hinfp−/−p53WT MEFs (conditional KO = cKO) and Hinfp−/−p53−/− MEFs (double knockout = dKO). The adenoviral vector used for Cre-recombinase expression co-expressed green fluorescent protein (GFP) marker that was used for live-cell selection. GFP positive cells expressing the Cre-recombinase were sorted 36 h after infection (BD FACS Aria I) and collected cells were grown in culture for up to 4–5 d (Fig. S1B). The GFP positive cells were genotyped post-sort to confirm removal of Hinfp (Fig. S1C). MEFs wild type for Hinfp and p53 were generated from original cHinfp colony whenever required. As described previously 24 the genomic ablation of Hinfp (cKO) generates truncated Hinfp gene transcripts. We also confirmed expression of truncated Hinfp and p53 transcripts in dKO cells (Fig. S1D and data not shown).
RNA extraction and qPCR
Total RNA was isolated from MEFs using the miRNeasy Mini Kit (QIAGEN Cat# 217004) followed by cDNA synthesis using the SuperScript First-Strand Synthesis System III (Invitrogen Cat# 18080–051). Relative transcript levels were determined by the ΔΔCt method using the cycle threshold (Ct) obtained in ViiA 7™ (Applied Biosystems by Life Technologies, Carlsbad, CA, USA) and iTaq Fast SYBR Green Supermix with ROX (Bio-Rad Laboratories Cat# 172–5122). Primer sequences are listed in supplementary table.
Immunofluorescence (IF)
GFP sorted MEFs (cKO and dKO) or WT or p53 null MEFs were plated on coverslips and cultured for 4 days; samples were collected at D1–D4. IF was carried out as described previously.15 Briefly, cells were fixed with 3.7% formaldehyde for 10 min at room temperature, permeabilized with 0.25% Triton X-100 for 20 min, and then incubated with primary antibody for 1h at 37°C, followed by detection using appropriate fluorescent-tagged secondary antibody. The nuclei were counterstained with DAPI. The following antibodies were used for protein detection: NPAT mouse monoclonal (1:1000; BD Transduction Laboratories Cat# 611344), α-tubulin mouse monoclonal (1:1000; Sigma Cat# T5168), γ-H2Ax-S139 mouse monoclonal (1:250; Millipore Cat# 05–636), 53BP1mouse monoclonal (1:250; Santa Cruz Biotechnology Cat# sc22760). Cells were viewed under an epifluorescence Zeiss AxioImager microscope equipped with a Hamamatsu charged coupled device (CCD) camera. Images were captured using 10×, 40×, 63×, or 100× objective magnification and Zen 2011 imaging software (Zeiss, Munich, Germany).
BrdU incorporation
WT, p53 null, cKO and dKO MEFs were grown either on coverslips (for IF) or in 6-well plates (for FACS). Incorporation of 5-bromo-2′-deoxyuridine (BrdU) (Roche Cat# 11296736001) was done by pulse labeling for 30 min at 37°C before harvesting each time point. Cells were then processed as per manufacturer's protocol for either detection by IF or Flow Cytometry (FACS). For IF, cells were fixed with ethanol and 50 mM glycine (pH 2.0) for 20 min at 20°C. BrdU signal was detected using fluorescent tagged antibodies and visualized by IF microscopy. Percentage of S-phase cells (BrdU positive) was determined by counting 200 nuclei per sample. For FACS analysis cells were processed using a BrdU – APC FACS kit (BD -PharMingen Cat# 51–9000019AK). Cells stained for BrdU and the DNA dye 7-AAD were analyzed using BD LSR II flow cytometer and cell cycle analysis was performed using FlowJo software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the members of our laboratory, especially Kristiaan Finstad for mouse colony maintenance, and others for helpful discussions and suggestions. We also thank Jennifer Díaz for assistance with manuscript preparation. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Funding
This work was supported by a National Institutes of Health grant R01 CA139322 to GSS and R01 CA077735 to SNJ.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Luger K. Dynamic nucleosomes. Chromosome Res 2006; 14:5-16; PMID: 16506092 [DOI] [PubMed] [Google Scholar]
- 2.Medina R, Ghule PN, Cruzat F, Barutcu AR, Montecino M, Stein JL, van Wijnen AJ, Stein GS. Epigenetic control of cell cycle-dependent histone gene expression is a principal component of the abbreviated pluripotent cell cycle. Mol Cell Biol 2012; 32:3860-71; PMID: 22826438; http://dx.doi.org/ 10.1128/MCB.00736-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hummon AB, Pitt JJ, Camps J, Emons G, Skube SB, Huppi K, Jones TL, Beissbarth T, Kramer F, Grade M, et al.. Systems-wide RNAi analysis of CASP8AP2/FLASH shows transcriptional deregulation of the replication-dependent histone genes and extensive effects on the transcriptome of colorectal cancer cells. Mol Cancer 2012; 11:1; PMID: 22216762; http://dx.doi.org/ 10.1186/1476-4598-11-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stein GS, van Wijnen AJ, Stein JL, Lian JB, Montecino M, Zaidi SK, Braastad C. An architectural perspective of cell-cycle control at the G1/S phase cell-cycle transition. J Cell Physiol 2006; 209:706-10; PMID: 17001681 [DOI] [PubMed] [Google Scholar]
- 5.Braastad CD, Hovhannisyan H, van Wijnen AJ, Stein JL, Stein GS. Functional characterization of a human histone gene cluster duplication. Gene 2004; 342:35-40; PMID: 15527963 [DOI] [PubMed] [Google Scholar]
- 6.Holmes WF, Braastad CD, Mitra P, Hampe C, Doenecke D, Albig W, Stein JL, van Wijnen AJ, Stein GS. Coordinate control and selective expression of the full complement of replication-dependent histone H4 genes in normal and cancer cells. J Biol Chem 2005; 280:37400-7; PMID: 16131487 [DOI] [PubMed] [Google Scholar]
- 7.Medina R, Buck T, Zaidi SK, Miele-Chamberland A, Lian JB, Stein JL, van Wijnen AJ, Stein GS. The histone gene cell cycle regulator HiNF-P is a unique zinc finger transcription factor with a novel conserved auxiliary DNA-binding motif. Biochemistry 2008; 47:11415-23; PMID: 18850719; http://dx.doi.org/ 10.1021/bi800961d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitra P, Xie RL, Medina R, Hovhannisyan H, Zaidi SK, Wei Y, Harper JW, Stein JL, van Wijnen AJ, Stein GS. Identification of HiNF-P, a key activator of cell cycle-controlled histone H4 genes at the onset of S phase. Mol Cell Biol 2003; 23:8110-23; PMID: 14585971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miele A, Braastad CD, Holmes WF, Mitra P, Medina R, Xie R, Zaidi SK, Ye X, Wei Y, Harper JW, et al.. HiNF-P directly links the cyclin E/CDK2/p220NPAT pathway to histone H4 gene regulation at the G1/S phase cell cycle transition. Mol Cell Biol 2005; 25:6140-53; PMID: 15988025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medina R, van Wijnen AJ, Stein GS, Stein JL. The histone gene transcription factor HiNF-P stabilizes its cell cycle regulatory co-activator p220NPAT. Biochemistry 2006; 45:15915-20; PMID: 17176114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao J, Dynlacht B, Imai T, Hori T, Harlow E. Expression of NPAT, a novel substrate of cyclin E-CDK2, promotes S-phase entry. Genes Dev 1998; 12:456-61; PMID: 9472014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao J, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA, Harlow E. NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes Dev 2000; 14:2283-97; PMID: 10995386 [PMC free article] [PubMed] [Google Scholar]
- 13.Ma T, Van Tine BA, Wei Y, Garrett MD, Nelson D, Adams PD, Wang J, Qin J, Chow LT, Harper JW. Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev 2000; 14:2298-313; PMID: 10995387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye X, Wei Y, Nalepa G, Harper JW. The cyclin E/Cdk2 substrate p220(NPAT) is required for S-phase entry, histone gene expression, and Cajal body maintenance in human somatic cells. Mol Cell Biol 2003; 23:8586-600; PMID: 14612403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghule PN, Becker KA, Harper JW, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Cell cycle dependent phosphorylation and subnuclear organization of the histone gene regulator p220(NPAT) in human embryonic stem cells. J Cell Physiol 2007; 213:9-17; PMID: 17520687 [DOI] [PubMed] [Google Scholar]
- 16.Ghule PN, Dominski Z, Yang XC, Marzluff WF, Becker KA, Harper JW, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Staged assembly of histone gene expression machinery at subnuclear foci in the abbreviated cell cycle of human embryonic stem cells. Proc Natl Acad Sci U S A 2008; 105:16964-9; PMID: 18957539; http://dx.doi.org/ 10.1073/pnas.0809273105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bongiorno-Borbone L, De Cola A, Vernole P, Finos L, Barcaroli D, Knight RA, Melino G, De Laurenzi V. FLASH and NPAT positive but not Coilin positive Cajal Bodies correlate with cell ploidy. Cell Cycle 2008; 7:2357-67; PMID: 18677100 [DOI] [PubMed] [Google Scholar]
- 18.Dominski Z, Marzluff WF. Formation of the 3′ end of histone mRNA: getting closer to the end. Gene 2007; 396:373-90; PMID: 17531405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker KA, Stein JL, Lian JB, van Wijnen AJ, Stein GS. Establishment of histone gene regulation and cell cycle checkpoint control in human embryonic stem cells. J Cell Physiol 2007; 210:517-26; PMID: 17096384 [DOI] [PubMed] [Google Scholar]
- 20.Gao G, Bracken AP, Burkard K, Pasini D, Classon M, Attwooll C, Sagara M, Imai T, Helin K, Zhao J. NPAT expression is regulated by E2F and is essential for cell cycle progression. Mol Cell Biol 2003; 23:2821-33; PMID: 12665581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie RL, Liu L, Mitra P, Stein JL, van Wijnen AJ, Stein GS. Transcriptional activation of the histone nuclear factor P (HiNF-P) gene by HiNF-P and its cyclin E/CDK2 responsive co-factor p220NPAT defines a novel autoregulatory loop at the G1/S phase transition. Gene 2007; 402:94-102; PMID: 17826007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie R, Medina R, Zhang Y, Hussain S, Colby J, Ghule P, Sundararajan S, Keeler M, Liu LJ, van der Deen M, et al.. The histone gene activator HINFP is a nonredundant cyclin E/CDK2 effector during early embryonic cell cycles. Proc Natl Acad Sci U S A 2009; 106:12359-64; PMID: 19590016; http://dx.doi.org/ 10.1073/pnas.0905651106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu LJ, Xie R, Hussain S, Lian JB, Rivera-Perez J, Jones SN, Stein JL, Stein GS, van Wijnen AJ. Functional coupling of transcription factor HiNF-P and histone H4 gene expression during pre- and post-natal mouse development. Gene 2011; 483:1-10; PMID: 21605641; http://dx.doi.org/ 10.1016/j.gene.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghule PN, Xie RL, Medina R, Colby JL, Jones SN, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Fidelity of histone gene regulation is obligatory for genome replication and stability. Mol Cell Biol 2014; 34:2650-9; PMID: 24797072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz D, Rotter V. p53-dependent cell cycle control: response to genotoxic stress. Semin Cancer Biol 1998; 8:325-36; PMID: 10101798 [DOI] [PubMed] [Google Scholar]
- 26.Hanel W, Moll UM. Links between mutant p53 and genomic instability. J Cell Biochem 2012; 113:433-9; PMID: 22006292; http://dx.doi.org/ 10.1002/jcb.23400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attardi LD, de Vries A, Jacks T. Activation of the p53-dependent G1 checkpoint response in mouse embryo fibroblasts depends on the specific DNA damage inducer. Oncogene 2004; 23:973-80; PMID: 14749764 [DOI] [PubMed] [Google Scholar]
- 28.Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ Jr.. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992; 71:587-97; PMID: 1423616 [DOI] [PubMed] [Google Scholar]
- 29.Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, Giovanella BC, Tainsky MA, Bradley A, Donehower LA. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene 1993; 8:2457-67; PMID: 8103211 [PubMed] [Google Scholar]
- 30.Jones SN, Sands AT, Hancock AR, Vogel H, Donehower LA, Linke SP, Wahl GM, Bradley A. The tumorigenic potential and cell growth characteristics of p53-deficient cells are equivalent in the presence or absence of Mdm2. Proc Natl Acad Sci U S A 1996; 93:14106-11; PMID: 8943068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cross SM, Sanchez CA, Morgan CA, Schimke MK, Ramel S, Idzerda RL, Raskind WH, Reid BJ. A p53-dependent mouse spindle checkpoint. Science 1995; 267:1353-6; PMID: 7871434 [DOI] [PubMed] [Google Scholar]
- 32.Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. Abnormal centrosome amplification in the absence of p53. Science 1996; 271:1744-7; PMID: 8596939 [DOI] [PubMed] [Google Scholar]
- 33.Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, et al.. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol 2002; 4:993-7; PMID: 12447390 [DOI] [PubMed] [Google Scholar]
- 34.Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol 2001; 21:1719-29; PMID: 11238909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358:15-6; PMID: 1614522 [DOI] [PubMed] [Google Scholar]
- 36.Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci U S A 1995; 92:8493-7; PMID: 7667317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Houtgraaf JH, Versmissen J, van der Giessen WJ. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc Revasc Med 2006; 7:165-72; PMID: 16945824 [DOI] [PubMed] [Google Scholar]
- 38.Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 2014; 13:217-36; PMID: 24577402; http://dx.doi.org/ 10.1038/nrd4236 [DOI] [PubMed] [Google Scholar]
- 39.Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nat Rev Clin Oncol 2011; 8:25-37; PMID: 20975744; http://dx.doi.org/ 10.1038/nrclinonc.2010.174 [DOI] [PubMed] [Google Scholar]
- 40.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402-12; PMID: 18431400; http://dx.doi.org/ 10.1038/nrm2395 [DOI] [PubMed] [Google Scholar]
- 41.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov 2008; 7:979-87; PMID: 19043449; http://dx.doi.org/ 10.1038/nrd2656 [DOI] [PubMed] [Google Scholar]
- 42.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356:215-21; PMID: 1552940 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.