

Abstract
Alavian and colleagues recently provided further evidence in support of the notion that the c subunit of the mitochondrial F1FO ATP synthase constitutes the long-sought pore-forming unit of the supramolecular complex responsible for the so-called ‘mitochondrial permeability transition’ (MPT). Besides shedding new light on the molecular mechanisms that underlie the MPT, these findings corroborate the notion that several components of the cell death machinery, including cytochrome c and the F1FO ATP synthase, mediate critical metabolic activities.
Keywords: apoptosis, BCL-XL, cyclophilin D, cyclosporin A, necrosis, permeability transition pore complex
Abbreviations
- AIFM1
apoptosis-inducing factor mitochondrion-associated, 1
- ATP5G
ATP synthase, H+ transporting, mitochondrial FO complex, subunit C
- CsA
cyclosporin A
- CYPD
cyclophilin D
- CYTC
holocytochrome c
- Δψm
mitochondrial transmembrane potential
- IMM
inner mitochondrial membrane
- MCU
mitochondrial calcium uniporter
- MPT
mitochondrial permeability transition
- PPIF
peptidylprolyl isomerase F
- PTPC
permeability transition pore complex
- RCD
regulated cell death
- SMV
submitochondrial vesicle.
The term ‘mitochondrial permeability transition’ (MPT) is generally employed to indicate an abrupt increase of the permeability of the inner mitochondrial membrane (IMM) to small solutes. This results not only in the immediate dissipation of the mitochondrial transmembrane potential (Δψm), and hence in the arrest of mitochondrial ATP synthesis, but also in a profound ionic imbalance that provokes the osmotic breakdown of the organelle.1 Accordingly, widespread MPT marks the point-of-no-return of several instances of regulated cell death (RCD), often (but not only) manifesting with biochemical and morphological correlates of necrosis.2-4 The MPT is caused by the opening of a relatively unselective multicomponent pore assembled at the junctions between the inner and outer mitochondrial membrane, the so-called ‘permeability transition pore complex’ (PTPC).5,6 The pharmacological profile of the PTPC has been intensively investigated throughout the past 2 decades.1,7 Nonetheless, its precise molecular composition, and in particular the identity of the pore-forming unit of the complex, remained a matter of debate until recently. Indeed, while a large number of studies pointed to various mitochondrial and cytosolic proteins as to structural or regulatory PTPC components, robust genetic evidence in support of these conclusions was lacking.8-10 As a standalone exception, peptidylprolyl isomerase F (PPIF), a protein of the mitochondrial matrix best known as cyclophilin D (CYPD), has been ascribed a key and non-redundant regulatory role in the MPT as early as in 2005.11-14
Based on several lines of circumstantial evidence, including pharmacological profile and interacting partners,7,15-17 we were among the first to hypothesize that the ATP synthasome, the multicomponent system that harnesses the electrochemical gradient generated across the IMM by the mitochondrial respiratory chain to produce ATP, is directly involved in the MPT.18-20 In particular, we were the first to demonstrate that the c subunit of FO (the IMM-embedded domain of the F1FO ATP synthase) is required for MPT, mitochondrial fragmentation and cell death as induced by mitochondrial Ca2+ overload and oxidative stress (two prototypic MPT inducers).18 Our observations were confirmed a few months later by an independent study, proving that isolated c subunits form pores in artificial membranes and that these pores are sensitive to established MPT regulators, including Ca2+.21 In spite of these findings, however, the possibility that c-rings (i.e., the ring-shaped oligomers of c subunits that – together with other proteins – make up the FO domain) would be available to generate MPT-like currents in cellula remained to be addressed.
The group of Elizabeth Jonas recently shed new light on the essential role played by the c subunit in MPT. By using reconstituted liposomes as well as purified mitochondria and submitochondrial vesicles (SMVs), Alavian and colleagues confirmed that c subunits can form multiconductance pores exhibiting a pharmacological profile that is similar to, but not perfectly overlapping with, that of the PTPC. On the one hand, the channel activity of highly purified c subunits reconstituted in liposomes as well as of SMVs exposed to urea (which denatures and removes extra-IMM proteins, including the F1 domain) was shown to be insensitive to both Ca2+ ions and to cyclosporin A (CsA), a prototypical CYPD inhibitor, contrary to that of untreated SMVs and whole mitochondria. On the other hand, c subunit-containing liposomes and urea-treated SMVs displayed a channel activity that could still be inhibited by ATP, although with a reduced efficacy.22 Taken together, these observations indicate that Ca2+ ions and CsA modulate the MPT by acting solely on extra-IMM components of the PTPC. Conversely, at least part of the MPT-inhibitory activity of ATP appears to reflects its ability to directly bind c-rings.
By means of the bipartite tetracysteine display method, Alavian et al. also demonstrated that CsA maintains c subunits close together in the IMM, as it protects human embryonic kidney HEK293 cells from the MPT-inducing effects of ionomycin (a ionophore that promotes mitochondrial Ca2+ accumulation). Conversely, control cells were shown to respond to ionomycin with a decrease in the relative proximity of c subunits. These data suggest that the MPT occurs in cellula along with a structural rearrangement of c-rings. Accordingly, the depletion of c subunits with short hairpin RNAs (shRNAs) specific for ATP synthase, H+ transporting, mitochondrial FO complex, subunit C1 (ATP5G1) or ATP5G3 (two of the three human isoforms of the c subunit) was found to resemble CsA in its ability to protect glycolytic cancer cells against MPT, Δψm dissipation and death as induced by ionomycin. Similar results were obtained with neurons maintained in normal medium and exposed to MPT-inducing agents such as H2O2 or glutamate. Conversely, the stable knockdown of ATP5G1 or ATP5G3 had a negative impact on the survival of HEK293 cells cultured in glucose-free, galactose-containing medium (which forces oxidative phosphorylation), an effect that could be rescued by means of a non-interferable c subunit-coding construct. Of note, a plasmid encoding a c subunit variant that contains 4 glycine-to-valine substitutions (increasing the steric hindrance of the protein and the conductance of c-rings) not only was unable to rescue the survival of HEK293 cells depleted of endogenous c subunits and maintained in these conditions, but also killed per se neurons cultured in normal medium in a CsA-insensitive manner.22 These data suggest that alterations in the ability of c subunits to pack tightly influence the efficacy of mitochondrial ATP synthesis as well as the propensity of c-rings to form unselective pores across the IMM in response to MPT-promoting stimuli.
Finally, Alavian and colleagues reported that purified β subunits (which are part of the F1 domain of the ATP synthase) inhibit the channel activity of purified c subunits reconstituted in liposomes. Taken together with the results obtained with urea-deprived SMVs, this observation suggests that the MPT may occur along with a physical dissociation between F1 and c-rings. Indeed, c subunits were released from the FOF1 ATP synthase in the presence of Ca2+ ions, a process that could be blocked by CsA as well by the absence of CYPD (i.e., in mitochondria isolated from Ppif−/- mice).22 These findings are compatible with a model proposing that the CYPD-dependent binding of Ca2+ ions to the F1 domain of ATP synthase displaces it from c-rings, hence initiating the MPT. Various components of the ATP synthasome, including solute carrier family 25, member 4 (SLC25A4), best known as adenine nucleotide translocase 1 and SLC25A3, best known as mitochondrial phosphate carrier, have been shown to alter the propensity of cells to undergo MPT-driven RCD,23,24 but the underlying molecular mechanisms remain to be elucidated (Figure 1).
Figure 1.
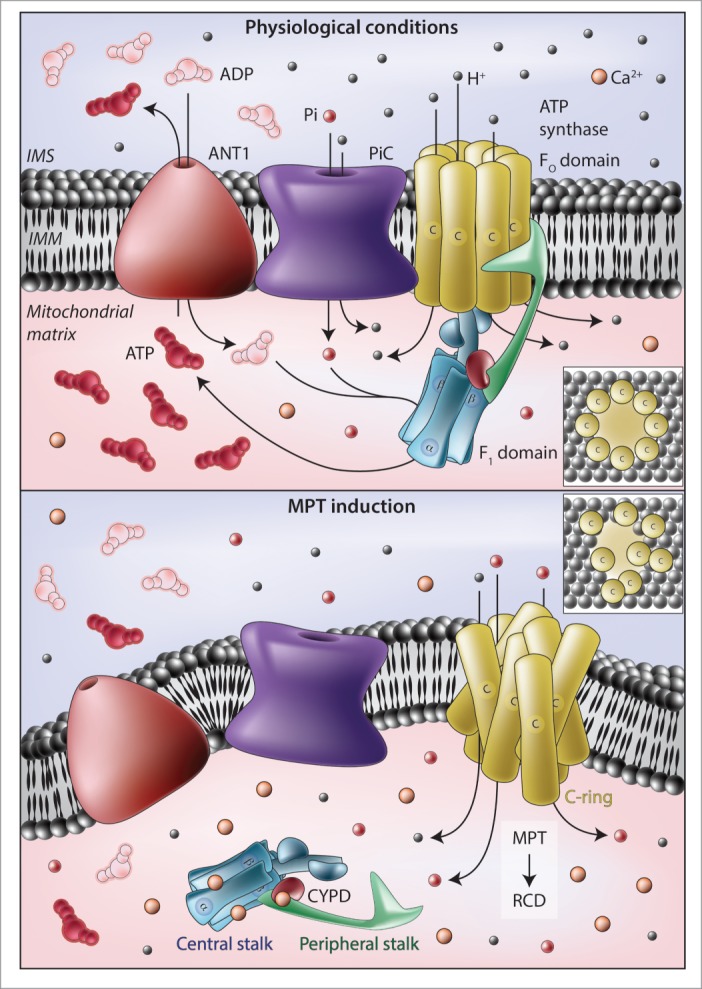
Implication of the ATP synthasome in the mitochondrial permeability transition. (A) In physiological conditions, the F1FO ATP synthase harnesses the electrochemical gradient established across the inner mitochondrial membrane (IMM) by respiratory chain complexes to produce ATP. The substrates of this reaction, i.e., ADP and inorganic phosphate (Pi), are provided by various members of the solute carrier (SLC) protein family, including SLC25A4, best known as adenine nucleotide translocase 1 (ANT1) and SLC25A3, best known as mitochondrial phosphate carrier (PiC). In particular, ANT1 exports ATP from the mitochondrial matrix in exchange of ADP (both of which are transported along their concentration gradient), whereas PiC operates as an H+-driven Pi/H+ symporter. (B) In response to oxidative stress or cytosolic Ca2+ overload, Ca2+ ions accumulate in the mitochondrial matrix and bind to the F1 domain of the F1FO ATP synthase, an activity that may be regulated by peptidylprolyl isomerase F (PPIF, best known as cyclophilin D, CYPD). In these conditions, F1 domains appear to dissociate from their IMM-embedded interacting partners (FO domains), allowing c-rings to structurally rearrange and form relatively unselective pores that initiate the mitochondrial permeability transition (MPT). Both ANT1 and PiC have been shown to influence the propensity of cells to undergo MPT-driven regulated cell death (RCD), but the precise molecular mechanisms remain elusive. IMS, mitochondrial intermembrane space.
In conclusion, the report by Alavian and collaborators provide novel insights into the molecular mechanisms that underlie MPT-driven RCD. Several issues, however, remain unresolved. First, it will be interesting to precisely determine how c-rings form high conductance pores. Indeed, the flow of H+ ions that powers ATP synthesis does not normally occur through the center of c-rings, which is highly hydrophobic, but at the interface between c-rings, other FO subunits and the IMM.25 Does the flow of small solutes that underpin the MPT occurs via the same route? Second, it will be important to understand how the association between FO and F1 ATP synthase domains is influenced by known regulators of the MPT, including metabolites, chemicals and PTPC interactors. Several members of the Bcl-2 protein family, including BCL2-like 1 (BCL2L1, best known as BCL-XL) and BCL2-associated X protein (BAX) have been shown to interact with components of the ATP synthasome, hence regulating their functions.17,24 Is the ability of these proteins to inhibit or promote MPT a direct consequence of such binding or does it reflect alterations in the local concentrations of ATP and ADP (both which are known MPT regulators)? Third, it will be crucial to characterize in detail the role of CYPD in the dissociation of F1 domains from c-rings and the consequent initiation of MPT. As mentioned above, CYPD currently represents the only PTPC component firmly established by genetic data, but does not constitute its pore-forming unit. Mitochondria lacking CYPD require markedly increased amounts of Ca2+ ions to undergo the MPT, but are not completely resistant to the process.12 Still, CYPD-deficient cells and mice are protected against a wide panel of MPT-triggering insults.11,13,14 Mcu−/− mitochondria, which are unable to rapidly take up cytosolic Ca2+ as they lack the mitochondrial calcium uniporter (MCU), also do not undergo the MPT in response to standard Ca2+ levels (500 μM). Mcu−/− cells and animals, however, are as sensitive to MPT inducers as their wild-type counterparts, yet fail to respond to CsA.26 How can these observations be reconciled into a single MPT model? Further experiments are required to clarify these points.
Irrespective of these incognita, the data by Alavian and colleagues lend further support to the notion that several factors involved in the initiation or execution of RCD exert key metabolic or bioenergetic functions.27,28 To mention two notable examples, this applies to holocytochrome c (CYTC) and apoptosis-inducing factor, mitochondrion-associated, 1 (AIFM1). Besides driving the assembly of the caspase-activating platform known as apoptosome upon mitochondrial outer membrane permeabilization (MOMP), CYTC shuttles electrons between respiratory complex III and IV, hence exerting a non-redundant vital function. Along similar lines, AIFM1 not only operates as a caspase-independent cell death executioner as it translocates to the nucleus upon MOMP or MPT and mediates large-scale DNA degradation, but also is required for the stability and function of respiratory complex I. Accordingly, both Cyc1−/− mice (lacking the protein moiety of CYTC) and Aifm1−/− mice do not survive through adulthood.29,30 This has profound implications for cell death research, de facto precluding the generation of appropriate genetic models. In some instances, such an issue can be (at least partially) circumvented by the establishment of tissue-specific knock-out animals.31 The gold standard approach to this problem would rely on the generation of mutant proteins that are unable to perform lethal functions but preserve their bioenergetic activities and homozygous knock-in mice. Such a strategy has been successfully undertaken by introducing a K72A substitution in the Cyc1-coding sequence, resulting in a CYTC variant that exerts normal respiratory functions but cannot bind apoptotic peptidase-activating factor 1 (APAF1) and hence cannot activate the caspase cascade.32 Unfortunately, not all RCD-relevant proteins might be amenable to such manipulation, for at least two reasons. First, it may not be possible to molecularly dissociate the vital and lethal activities of all proteins involved in the initiation or execution of RCD by mutagenesis. Second, the existence of a relatively consistent degree of genetic and epigenetic redundancy may prevent (or at least complicate significantly) the establishment of bona fide knock-in models. This latter issue applies to the c subunit of the FOF1 ATP synthase, which in the mouse genome is coded by 3 distinct loci (Atp5g1, Atp5g2 and Atp5g3).33 It may therefore be difficult to obtain unequivocal genetic evidence demonstrating the true pathophysiological relevance of c-rings for MPT-driven RCD in vivo. Irrespective of these obstacles, it is now clear that the ATP synthasome occupies a central position in the molecular mechanisms that regulate the transition between a cell's life and its death.
Funding Statement
MB and PP are supported by the Italian Association for Cancer Research (AIRC); Telethon (GGP11139B); local funds from the University of Ferrara; the Italian Ministry of Education, University and Research (COFIN, FIRB, and Futuro in Ricerca); and an Italian Ministry of Health grant. GK and LG are supported by the Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR); Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Fondation Bettencourt-Schueller; Fondation de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
References
- 1. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87:99-163; PMID:17237344; http://dx.doi.org/ 10.1152/physrev.00013.2006 [DOI] [PubMed] [Google Scholar]
- 2. Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol 2014; PMID:24582829; http://dx.doi.org/ 10.1016/j.semcdb.2014.02.006 [DOI] [PubMed] [Google Scholar]
- 3. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19:107-20; PMID:21760595; http://dx.doi.org/ 10.1038/cdd.2011.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16:3-11; PMID:18846107; http://dx.doi.org/ 10.1038/cdd.2008.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brenner C, Moulin M. Physiological roles of the permeability transition pore. Circ Res 2012; 111:1237-47; PMID:23065346; http://dx.doi.org/ 10.1161/CIRCRESAHA.112.265942 [DOI] [PubMed] [Google Scholar]
- 6. Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 2009; 46:821-31; PMID:19265700; http://dx.doi.org/ 10.1016/j.yjmcc.2009.02.021 [DOI] [PubMed] [Google Scholar]
- 7. Bonora M, Wieckowski MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L, Pinton P. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 2014; 0; PMID:24727893; http://dx.doi.org/ 10.1038/onc.2014.96 [DOI] [PubMed] [Google Scholar]
- 8. Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004; 427:461-5; PMID:14749836; http://dx.doi.org/ 10.1038/nature02229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Galluzzi L, Kroemer G. Mitochondrial apoptosis without VDAC. Nat Cell Biol 2007; 9:487-9; PMID:17473857; http://dx.doi.org/ 10.1038/ncb0507-487 [DOI] [PubMed] [Google Scholar]
- 10. Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 2007; 9:550-5; PMID:17417626; http://dx.doi.org/ 10.1038/ncb1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005; 434:658-62; PMID:15800627; http://dx.doi.org/ 10.1038/nature03434 [DOI] [PubMed] [Google Scholar]
- 12. Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem 2005; 280:18558-61; PMID:15792954; http://dx.doi.org/ 10.1074/jbc.C500089200 [DOI] [PubMed] [Google Scholar]
- 13. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005; 434:652-8; PMID:15800626; http://dx.doi.org/ 10.1038/nature03317 [DOI] [PubMed] [Google Scholar]
- 14. Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A 2005; 102:12005-10; PMID:16103352; http://dx.doi.org/ 10.1073/pnas.0505294102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McGeoch JE, Palmer DN. Ion pores made of mitochondrial ATP synthase subunit c in the neuronal plasma membrane and Batten disease. Mol Genet Metab 1999; 66:387-92; PMID:10191134; http://dx.doi.org/ 10.1006/mgme.1999.2822 [DOI] [PubMed] [Google Scholar]
- 16. Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem 2009; 284:33982-8; PMID:19801635; http://dx.doi.org/ 10.1074/jbc.M109.020115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol 2011; 13:1224-33; PMID:21926988; http://dx.doi.org/ 10.1038/ncb2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013; 12:674-83; PMID:23343770; http://dx.doi.org/ 10.4161/cc.23599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA 2013; 110:5887-92; PMID:23530243; http://dx.doi.org/ 10.1073/pnas.1217823110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Marchi E, Bonora M, Giorgi C, Pinton P. The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium 2014; 56:1-13; PMID:24755650; http://dx.doi.org/ 10.1016/j.ceca.2014.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Azarashvili T, Odinokova I, Bakunts A, Ternovsky V, Krestinina O, Tyynela J, Saris NE. Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 2014; 55:69-77; PMID:24380588; http://dx.doi.org/ 10.1016/j.ceca.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 22. Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA Jr, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci USA 2014; 111(29):10580-5; 2014:IN PRESS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alcala S, Klee M, Fernandez J, Fleischer A, Pimentel-Muinos FX. A high-throughput screening for mammalian cell death effectors identifies the mitochondrial phosphate carrier as a regulator of cytochrome c release. Oncogene 2008; 27:44-54; PMID:17621274; http://dx.doi.org/ 10.1038/sj.onc.1210600 [DOI] [PubMed] [Google Scholar]
- 24. Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prévost MC, Xie Z, Matsuyama S, Reed JC, et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 1998; 281:2027-31; PMID:9748162; http://dx.doi.org/ 10.1126/science.281.5385.2027 [DOI] [PubMed] [Google Scholar]
- 25. Symersky J, Pagadala V, Osowski D, Krah A, Meier T, Faraldo-Gomez JD, Mueller DM. Structure of the c(10) ring of the yeast mitochondrial ATP synthase in the open conformation. Nat Struct Mol Biol 2012; 19:485-91, S1; PMID:22504883; http://dx.doi.org/ 10.1038/nsmb.2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 2013; 15:1464-72; PMID:24212091; http://dx.doi.org/ 10.1038/ncb2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Galluzzi L, Joza N, Tasdemir E, Maiuri MC, Hengartner M, Abrams JM, Tavernarakis N, Penninger J, Madeo F, Kroemer G. No death without life: vital functions of apoptotic effectors. Cell Death Differ 2008; 15:1113-23; PMID:18309324; http://dx.doi.org/ 10.1038/cdd.2008.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep 2012; 13:322-30; PMID:22402666; http://dx.doi.org/ 10.1038/embor.2012.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown D, Yu BD, Joza N, Benit P, Meneses J, Firpo M, Rustin P, Penninger JM, Martin GR. Loss of Aif function causes cell death in the mouse embryo, but the temporal progression of patterning is normal. Proc Natl Acad Sci U S A 2006; 103:9918-23; PMID:16788063; http://dx.doi.org/ 10.1073/pnas.0603950103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, Wang X, Williams RS. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell 2000; 101:389-99; PMID:10830166; http://dx.doi.org/ 10.1016/S0092-8674(00)80849-1 [DOI] [PubMed] [Google Scholar]
- 31. Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 2007; 131:476-91; PMID:17981116; http://dx.doi.org/ 10.1016/j.cell.2007.08.047 [DOI] [PubMed] [Google Scholar]
- 32. Hao Z, Duncan GS, Chang CC, Elia A, Fang M, Wakeham A, Okada H, Calzascia T, Jang Y, You-Ten A, et al. Specific ablation of the apoptotic functions of cytochrome C reveals a differential requirement for cytochrome C and Apaf-1 in apoptosis. Cell 2005; 121:579-91; PMID:15907471; http://dx.doi.org/ 10.1016/j.cell.2005.03.016 [DOI] [PubMed] [Google Scholar]
- 33. http://www.ncbi.nlm.nih.gov/gene [Google Scholar]