

Abstract
Background/Aims
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide, and it has a poor prognosis and few therapeutic options. Radiotherapy is one of the most effective forms of cancer treatment, and P53 protein is one of the key molecules determining how a cell responds to radiotherapy. The aim of this study was to determine the therapeutic efficacy of iodine-131 in three human HCC cell lines.
Methods
Western blotting was used to measure P53 expression. The effects of radiotherapy with iodine-131 were assessed by using the clonogenic assay to evaluate cell survival. Flow cytometry was carried out to examine the effects of iodine-131 on cell death, oxidative stress, reduced intracellular glutathione expression, the mitochondrial membrane potential, and the cell cycle.
Results
The P53 protein was not expressed in Hep3B2.1-7 cells, was expressed at normal levels in HepG2 cells, and was overexpressed in HuH7 cells. P53 expression in the HuH7 and HepG2 cell lines increased after internal and external irradiation with iodine-131. Irradiation induced a decrease in cell survival and led to a decrease in cell viability in all of the cell lines studied, accompanied by cell death via late apoptosis/necrosis and necrosis. Irradiation with 131-iodine induced mostly cell-cycle arrest in the G0/G1 phase.
Conclusions
These results suggest that P53 plays a key role in the radiotherapy response of HCC.
Keywords: Hepatocellular carcinoma, Iodine-131, Radiotherapy, P53
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common primary liver malignancy with a rising incidence worldwide, being the second cause of cancer death.1 The prognosis of HCC patients is poor, and therapies are in most cases only the palliative approach. Surgical treatments have more satisfactory results, however, few patients can benefit from it. The therapeutic options for HCC often have more limitations and disappointing results.2,3 In this context, it is urgent to study and develop new therapeutic strategies.
Radiotherapy is one of the most effective anticancer therapies and it has been used to treat a wide variety of tumors. The success of radiotherapy against cancer depends on its ability to kill tumor cells, while preserving the adjacent normal tissue.4,5 Radiotherapy can be external and internal. In external radiotherapy, the device directs rays of high energy to the tumor tissue affecting also a small volume of surrounding normal tissue. In internal radiotherapy, the radiation source is placed in/or near the tumor site emitting radiation continuously for weeks, months or even one year, allowing an higher total dose of radiation to be applied in a shorter time than in the external radiotherapy.4,6,7 Several types of radiation sources can be used. One of them is beta radiation from different radionuclides.6,7
When, in 1943, radio-iodine was first used in the treatment of metastasized thyroid carcinoma, its success in tumor response was considered a "miracle". Since then, many efforts have been made to apply radioisotope therapy in other tumors.8 However, only a small number of radionuclides have been used to the HCC treatment, such as iodine-131, yttrium-90, rhenium-188 and holmium-166 with promising results.9,10,11
Iodine-131 is a radionuclide, with interesting properties for anticancer therapy. It is available with high radiochemical purity and specific activity, has a long half-life of 8.06 days and decays with production of beta particles (maximum energy of 606 keV), and gamma emissions (main peak at 364 keV).12
In radiotherapy there are a variety of cellular processes involving essential proteins, and which determine the success of treatment. The P53 protein is a transcription factor associated with more than 50% of human tumors and is a key molecule involved in the response to radiotherapy.13,14 It is encoded by the tumor suppressor gene TP53 located on the short arm of chromosome 17 (17p13.1 locus) and has 393 amino acids and four domains.14,15 The role of the tumor suppressor genes is to help in the prevention and repair of genetic damage. When cell damage occurs in the DNA, and P53 is functional, this protein may induce growth arrest, often in G1 or G2 phases of cell cycle, or lead to cell suicide by apoptosis, preventing uncontrolled proliferation and tumor development.4,5,13,16 In contrast, if the P53 protein is inactive, which can happen by mutation or degradation, DNA damage is not detected which leads to a genetic instability. This facilitates the initiation and progression of tumors and besides the tumors become resistant to chemo and radiotherapy.4,13,14 The resistance to radiation appears to be related to germ line P53 mutation, however, the association between P53 expression and radiotherapy response in HCC has not been well studied. Thus, the significance of P53 aberration in HCC remains inconclusive.4,5,13
This work aims to investigate the efficiency of iodine-131 radiotherapy in three HCC cell lines, and the influence of the P53 protein in the biological mechanisms of HCC. Three human HCC cell lines were exposed to different doses of internal or external radiation sources of iodine-131, which differ in P53 expression 17 and their viability, cell death and reactive oxygen species (ROS) production were as well evaluated.
MATERIAL AND METHODS
Cell culture
We used three human HCC cell lines: HepG2, HuH7 and Hep3B2.1-7. HepG2 and Hep3B2.1-7 cell lines were obtained from American Tissue Cell Collection (ATCC), USA. HuH7 cell line was obtained from Japanese Collection of Research Bioresources (JCRB), Japan. Cells were propagated on adherent cultures in Dulbecco's Modified Eagle Medium (DMEM) with 10% Fetal Bovine Serum (FBS) (Gibco 2010-09), 100 units/mL of penicillin and 100 µg/mL of streptomycin (Gibco 15140-122), and incubated at 37℃ in 5% CO2 atmosphere.
Irradiation
During the cells exponential growth phase, 5×105 cells were exposed to internal and external radiation with iodine-131 (IBA Molecular). The conditions used for each irradiation type are outlined in Tables 1 and 2. Concerning the internal radiation, the radionuclide was placed directly in contact with the cells that were washed afterwards with phosphate buffered saline (PBS). Related to the external radiation, a radionuclide source was positioned below the flask containing the cells. Doses were calculated assuming the worst case scenario, i.e., all the emitted energy in the decay process was absorbed by the cells. To calculate the internal radiation exposure dose, was used the equation 1,
| (equation 1) |
where D is the absorbed dose (Gy), A0 the initial activity of the radioactive source (mCi), T1/2 the half-life time (s), t the irradiation time (s), Ē the average energy per disintegration (eV) and M the mass of the sample subjected to irradiation (kg).
Table 1. Internal irradiation conditions.
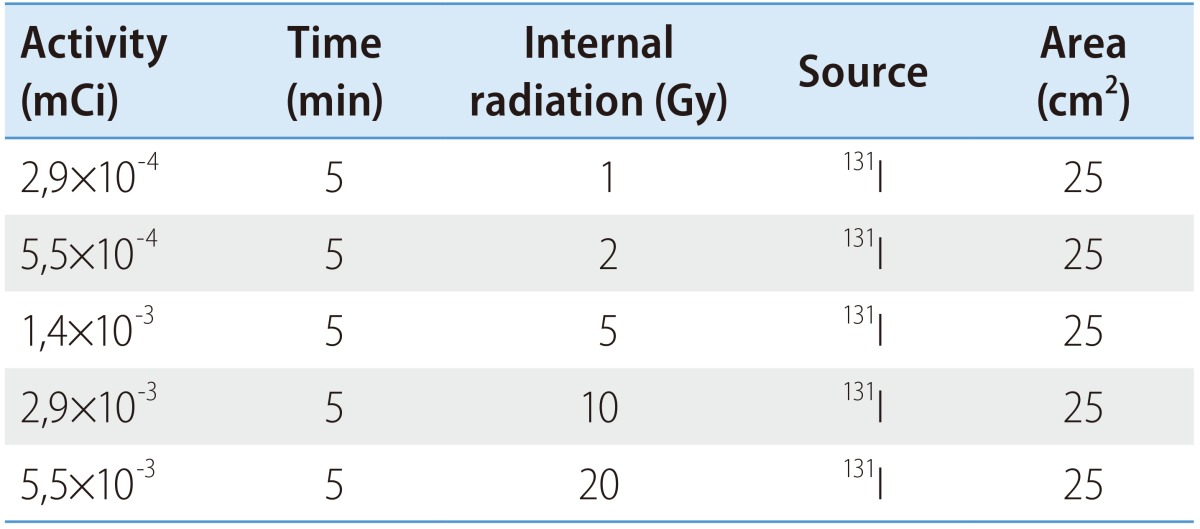
Table 2. External irradiation conditions.
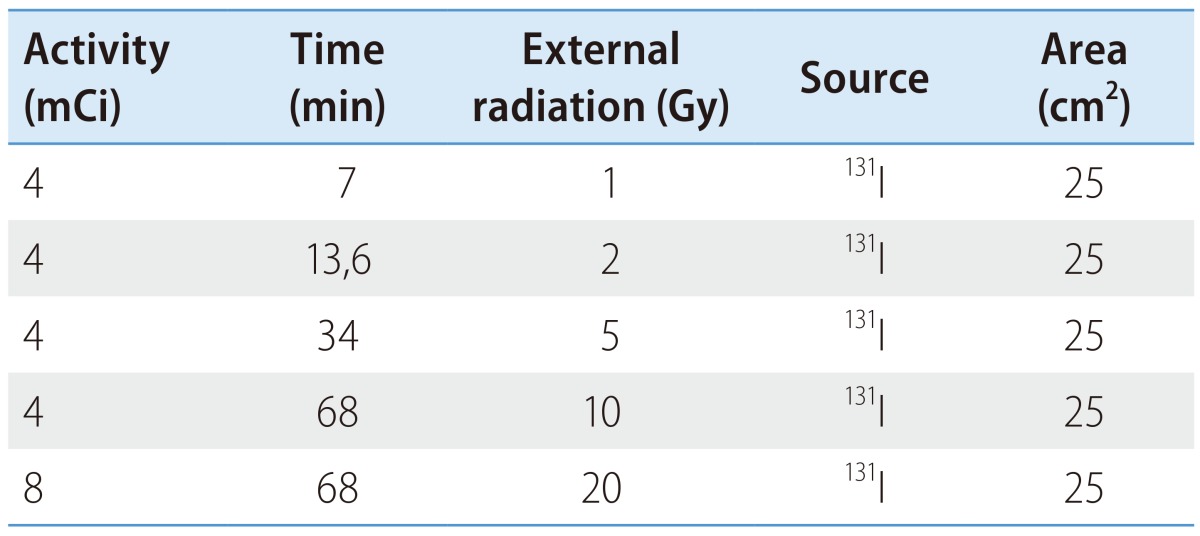
To calculate the external radiation exposure dose, was used the equation 2, where D is the exposure dose (mGy), Г radiation constant specifies , A the radioactive source activity (GBq), t the exposure time (h) and d the distance from the source (m).
| (equation 2) |
P53 and phosphorylated P53 expression
P53 and phosphorylated P53 protein (p-P53) protein expression levels were determined by western blot. Cell extracts were prepared in ice using a solution of radioimmunoprecipitation assay (RIPA) buffer and complete Mini Ethylenediamine tetraacetic acid (EDTA)-free (Roche). Protein concentrations were determined by bicinchoninic (BCA) method. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was held using a 10% acrylmide gel. Proteins were electrotransfered to nitrocellulose membrane (PVDF) at 4℃, during 1 hour. Membrane blocking was performed with 4% bovine serum albumin (BSA) in tris-buffered saline tween-20 (TBS-T) for 1 hour at room temperature. Membranes were incubated with primary antibody (P53 or p-P53 or β-actin, from Santa Cruz Biotechnology) and secondary antibody (mouse antibody, from Sigma) according to the manufacturer's instructions. The blots were stained with elemental chlorine free (ECF, Sigma) and read in 9000 Typhoon FLA equipment.
Cell survival
Cell survival was evaluated using clonogenic assay. Six hundred plated cells were irradiated with internal or external source of iodine-131 and twelve days afterwards were fixed with methanol and stained with crystal violet. Colonies with more than 50 cells were counted and the efficiency plate (EP) (relation between the number of colonies counted and the number of cells plated) and survival factor (SF) (relationship between EP of the treated sample and the EP of the control) were determined.
Flow cytometry
Flow cytometry was used to analyze cell viability, cell death and cell cycle as well as to determine ROS and reduced glutathione production and mitochondrial membrane potential. All flow cytometry studies used 106 cells per probe and exposures to internal or external radiation of 1 or 20 Gy occurred 24 h before the assay. For all procedures were performed controls, i.e. cells not subject to irradiation.
Cell viability and cell death
The influence of irradiation with iodine-131 on cell viability, and the types of induced cell death were determined by flow cytometry using annexin-V/propidium iodide (AV/PI) (KIT Immunotech). One of the main features of cell death by apoptosis is the redistribution of plasma membrane phosphatidylserine, a phospholipid that, in apoptotic cells, is translocated from the inner to the outer leaflet of the plasmatic membrane and binds to AV. Complementarily, PI, which does not permeate viable cells, binds to deoxyribonucleic acid (DNA) intercalating between the bases on late apoptotic and necrotic cells.18 In this assay, 106 cells were incubated during 15 min in binding buffer with 1 µL of AV (Kit Immunotech) and 5 µL of PI (Kit Immunotech). Subsequently, cells were excited at a wavelength of 525 nm for AV and 640 nm for PI, collecting 104 events to assess the percentage of viable, early apoptotic, late apoptotic/necrotic, and necrotic cells.18
Evaluation of ROS and reduced glutathione production, mitochondrial membrane potential and cell cycle
ROS production: intracellular peroxide were measured using 2',7'-dichlorodihydrofluorescein diacetate (DCFH2-DA, Invitrogen); intracellular superoxide was determined using the dihydroethidium probe (DHE, Sigma). Expression of intracellular reduced glutathione was evaluated with mercury orange (GSH, Sigma). Mitochondrial membrane potential was analyzed with 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazol carbocyanine (JC-1, Invitrogen). Cell cycle was evaluated with PI/RNase solution (Immunostep). Assays were performed according to a method described by Mamede et al.18 Results are presented as mean fluorescence intensity (MFI).
Statistical analysis
Cell survival curves were obtained using Origin Pro v8.0, fitting the experimental data to two models: for lower doses, the data was fitted to a linear-quadratic model, according to the equation SF=e-αD-βD2; for higher radiation doses, we fitted the data to a linear model, using SF=e-C-αD. These two models were chosen as they fitted the data when used together. P53, p-P53 expression, and all flow cytometry results were analyzed using IBM SPSS® v.19 (IBM Corporation, Armonk, New York, USA). In the descriptive analysis measures of central tendency (mean and median) and dispersion (standard deviation and interquartile range) were obtained for quantitative variables. To assess normality of the distribution of quantitative variables, Shapiro-Wilk's test was used. Parametric tests were used for comparisons in the case normality was observed; in the opposite case we used nonparametric tests. For comparison of quantitative variables between two groups we used Student's t test (parametric) or Mann-Whitney's test (nonparametric). For more than two groups we used one factor Analysis of Variance (ANOVA) test, with post-hoc Tukey test (parametric) or Kruskal-Wallis test with multiple comparisons performed using the Mann-Whitney's test with Bonferroni correction (nonparametric). For all comparisons a significance level of 5% was considered.
RESULTS
P53 and phosphorylated P53 expression
Basal P53 expression differs significantly across the three HCC cell lines studied (Fig. 1). P53 expression, as determined by western blotting, demonstrated to be highest in HuH7 cell line (0.48%), and null in Hep3B2.1-7 cells (0%). When HuH7 and HepG2 cell lines were exposed to the treatment with iodine-131, it was observed a gradual increase in P53 expression with the dose and with the time after irradiation as demonstrated in Figure 2. The HuH7 cells showed the highest levels of P53 expression (P<0.05), regardless of the dose and exposure duration of radiation, in comparison to HepG2 cell line. The P53 expression was higher, 24 hours after exposure at 20 Gy, both for internal or external radiation (P<0.05). For all doses and exposure duration tested, the levels of P53 protein expression were higher after external radiation.
Figure 1. Evaluation of basal P53 protein expression. Bands corresponding to P53 (53 kDa) and actin protein (47 kDa) in the HepG2, HuH7, and Hep3B2.1-7 cell lines. Data are mean and SE values (n=6). Hashes indicate statistically significant differences between the conditions (P<0.05).

Figure 2. Evaluation of P53 protein expressions in HepG2 and HuH7 cell lines in response to internal irradiation (IR) and external irradiation (ER) with different doses of iodine-131 and for different exposure durations. Data are mean and SE values (n=5). Asterisks indicate statistically significant differences compared to controls (P<0.05).

Phosphorylated P53 expression was present in all cell lines exposed to iodine-131 at 20 Gy for 24 hours, being higher after external irradiation compared to internal radiation (Fig. 3). HepG2 cells showed the highest levels of p-P53, with 1.99% of protein expression (P=0.002).
Figure 3. Evaluation of phosphorylated P53 protein expressions in HepG2 and HuH7 cell lines in response to IR and ER with different doses of iodine-131 and for different exposure durations. Data are mean and SE values (n=3). Asterisks indicate statistically significant differences compared to controls (P<0.05).

Cell survival, cell viability and cell death
The increasing exposure to radiation doses, led to a reduction in cell survival as represented in Figure 4. The HuH7 cells were the most radiosensitive to the internal irradiation for high doses, and HepG2 cells to the external irradiation, showing a greater reduction of cell survival. The Hep3B2.1-7 cells were the less radiosensitive to the both irradiation types.
Figure 4. Cell survival curves for HepG2, HuH7, and Hep3B2.1-7 cell lines in response to IR and ER with iodine-131. Data are mean and SD values (n=3).

Using internal irradiation, HepG2 cells showed 49% of viability (P=0.031), and a tendency to activate cell death by necrosis (P=0.03) at 20 Gy (Fig. 5). The HuH7 cells had 59% of viability after 1 Gy of irradiation, and 20% of cells died by apoptosis (P=0.02). The Hep3B2.1-7 cells had 49% of viability, and 24% died by apoptosis (P=0.003), after internal irradiation with 20 Gy. After 1 Gy external irradiation, the HepG2 cells had 40% of viability (P=0.013), and 28% of viability at 20 Gy (P=0.001). Regarding cell death, 32% of Hep3B2.1-7 cells died by necrosis with 1 Gy irradiation dose (P=0.017), and at 20 Gy, 25% died by late apoptosis/necrosis (P=0.016), and 35% by necrosis (P=0.031) (Fig. 5). The HuH7 cells showed 17% of death by late apoptosis/necrosis at 1 Gy and 20% at 20 Gy. The Hep3B2.1-7 cells had 49% of viability (P=0.036), 19% dying by late apoptosis/necrosis (P=0.024) and 20% by necrosis (P=0.046) at 20 Gy.
Figure 5. Cell viability analysis using flow cytometry with AnV/IP double staining. The results are percentages of cells that are viable, in apoptosis, in late apoptosis/necrosis, and in necrosis at 24 hours after IR or ER with iodine-131. Data are mean and SE values (n=8). Asterisks indicate statistically significant differences compared to controls (P<0.05).

ROS and reduced glutathione production, mitochondrial membrane potential and cell cycle
Regarding to the intracellular peroxide concentrations in cells exposed to internal 1 Gy irradiation was observed an increase in intracellular peroxide concentration in Hep3B2.1-7 cell line (P=0.015), relative to control cell population (Fig. 6A). Similarly, in the external irradiation, it was observed, for the same irradiation dose, an increase in intracellular peroxide concentration in HuH7 cells (P=0.013) as well as for 20 Gy (P=0.016), in comparison to control. The intracellular reduced glutathione concentration exposed to 1Gy external irradiation, increased in Hep3B2.1-7 cells (P=0.016), in comparison to control.
Figure 6. (a) Flow cytometry analysis of the production of intracellular peroxides and superoxide radicals, and of reduced intracellular glutathione expression and the mitochondrial membrane potential. Data are mean and SE values (n=8). Asterisks and hashes respectively indicate statistically significant differences compared to controls and between IR and ER (P<0.05). (b) Cell-cycle analysis in the HepG2, HuH7, and Hep3B2.1-7 cell lines by flow cytometry at 24 h after IR and ER exposure to iodine-131 at 1 to 20 Gy. Data are mean and SE values (n=8).

Comparing both irradiation types, there was a general tendency for a higher increase of ROS production especially in intracellular peroxide concentrations in HuH7 for 1 Gy (P=0.018) and 20 Gy (P=0.021) irradiation doses, and in superoxide radical concentration in HepG2 only for 1 Gy irradiation dose (P=0.046). Concerning glutathione production it was observed a decrease especially in Hep3B2.1-7 cell line after 1 Gy irradiation dose (P=0,027) as well as for mitochondrial membrane potential in which it was observed a decreased comparing the external with the internal irradiation results (Fig. 6A)
The results obtained in the cell cycle study showed that, compared with the control and regardless of cell line, dose and irradiation type used, the cell cycle was mostly arrested in G0/G1 phase (Fig. 6B). Moreover, arrest in G0/G1 phase was more evident in cells externally irradiated than in cells internally irradiated, for HuH7 and Hep3B2.1-7 cells. The HuH7 cells were the most sensitive.
DISCUSSION
Worldwide, HCC is the second leading cause of cancer-related death.1 Overall survival at 5 years for patients with HCC is approximately 2-10%, so it is urgent to develop new therapeutic strategies for this highly aggressive cancer.3 Radiotherapy is one of the most effective forms of cancer treatment and P53 is a key molecule involved in cellular response to ionizing radiation.5,13,14,19
Given the importance of P53 expression in the development and response to treatment of many types of tumors, and in order to determine the effectiveness of radiotherapy in different HCC cell lines, we evaluated the P53 expression protein in three HCC cell lines. One of the biochemical features which differ between these cell lines is the P53 expression levels. Thus, in response to radiotherapy the cell lines used showed differences in P53 protein expression. HuH7 and HepG2 had a basal P53 protein expression. The Hep3B2.1-7 cell line, which has a homozygous deletion in exon 11 of the TP53 gene, does not have the ability to express P53 protein.20,21,22,23,24,25,26,27,28,29,30,31 HepG2 cells, express P53 at intermediate level comparatively to HuH7 and Hep3B2.1-7 cell lines, as reported in literature.21,23,26,31 As expected and also described in literature by Reiser et al.26 and Bressac et al.,23 mutated P53 at codon 220 cystyr of exon 6 in HuH7 cells, induces an overexpression of this protein.22,25 Hep3B2.1-7 cell line did not express P53, and thus the expression of this protein in response to irradiation was not evaluated. But after internal and external radiation exposure, differences in P53 expression in HepG2 and HuH7 cells were observed. When comparing the P53 protein expression levels in HepG2 and HuH7 cell lines, using internal or external irradiation, the cell line that showed higher P53 expression was HuH7. Thus, we can confirm the P53 protein overexpression in HuH7 cells, as described in the literature.23,26 It was also found that, independently of cell line, for all doses and exposure duration, P53 protein expression was higher in external comparing with the radiation internal exposure.
In order to confirm if P53 protein was active after irradiation in HepG2 and HuH7 cell lines, phosphorylated P53 expression was evaluated for 24 hours exposure duration and 20 Gy dose. It was verified that the P53 protein was active in both cell lines, but more active when exposed to external radiation than internal radiation, which is coincident with P53 expression. Thus, the cell lines exposed to external radiation have a greater ability to repair cellular damage.
After verifying that the P53 expression was largely affected by the exposure to iodine-131, cell survival and cell viability were evaluated. As described in the literature by Gudkov and Komarova,4 "...P53 can be a determinant of radiosensitivity". The differences in radiosensitivity between cell lines are due the different levels of P53 expression: the most radiosensitive cell lines were those that exhibited a higher expression of P53, which is supported by a greater decrease on cell survival (HepG2 e HuH7). Allowing them, a greater ability to recognize cellular damage, than Hep3b2.1-7 cells.4,5,14 So, cell survival after treatment with iodine-131 might be influenced by P53 expression. The internal radiation is a little more aggressive than the external radiation, likely due to the direct exposure of cells to beta particles and gamma rays, while in the external radiation, the cells were exposed only to gamma rays.
It was expected that P53 protein triggered mostly apoptotic cell death.4,5,16,32,33 However, HepG2, HuH7 and Hep3B2.1-7 cells died by late apoptosis/necrosis and necrosis.34 Necrosis is triggered as a response to high dose radiation. Therefore, we must take into account the aggressiveness of radiation to which cells were subjected in the treatment with iodine-131, and the studies were performed 24 hours after treatment. Moreover, we cannot exclude the possibility that we observed a late stage in cell death pathway, which could initially or earlier have been apoptosis or autophagy or mitotic death.35
Through the observation of Figure 5, it appears that the HepG2 cell line is more sensitive to both the treatments used (internal and external irradiation) than HuH7 cell line. This result may be related to the fact that the HepG2 cell line express the normal and functional form of P53,17 thereby recognizing the damage caused and triggering a cell death process. In fact it is widely recognized the key role that P53 plays in the process of apoptosis, however, some studies have shown that P53 also has a leading role in necrosis,35,36 which reinforces the obtained results.
ROS are involved in a variety of cellular processes, including cell growth arrest or cell death. ROS are mainly produced in the body by exposure to ionizing radiation, and high concentrations of ROS cause cell death preferably by necrosis.38,39,40 In this study it was found some necrotic cell death and high levels of cell growth arrest.38,39,40
As expected and described in the literature by Gudkov and Komarova,4 Begg et al.5 and Levine and Oren,13 the phase of cell cycle in which there was higher cell growth arrest was the G0/G1 phase, being more evident for the highest dose (20 Gy), since very high doses induce higher activation of the P53 protein, responsible for cell growth arrest.4,5,13,14,16 It should also be observed that the levels of cell growth arrest were higher with external radiation, which was likely due to the aggressiveness of direct exposure of cells to beta particles and gamma rays, which causes additional cell stress and prohibits activation of the P53 protein.14,41
Reported by several researchers as a key molecule involved in cell response to ionizing radiation, the study of P53 protein in this experimental work was of high importance.4,5,13,14 Coincident with various studies one of main findings is that cells expressing P53 in larger quantities were the most radiosensitive (decreased viability), which is consistent with previously published reports on P53 protein expression in response to radiotherapy. This may implicate a more favorable prognosis for tumors which express this gene.4,5,14 Thus, we conclude that the TP53 tumor suppressor might be a key factor in response to radiotherapy.5,13 Another important message from this work is that the molecular profile of tumors should be explored in great detail, to try to predict the response of tumor cells to various therapeutic approaches. The signaling pathways involved in each mechanism of tumor development must be investigated, because the assessment of one protein does not provide a thorough understanding of the mechanisms of cellular response and, therefore, further investigation should be performed. The P53 mutations are one of the most significant alterations in cell regulation, which can lead to the development of HCC. Thus, we believe that this research is important for a better evaluation of clinical prognostic and development of therapeutic strategies that ensure a better patient survival.
Acknowledgements
Ana F Brito would like to thank the Portuguese Foundation for Science and Technology for the award of a PhD scholarship (SFRH/BD/61378/2009).
Mafalda Laranjo would like to thank the Portuguese Foundation for Science and Technology for the award of a PhD scholarship (SFRH/BD/44957/2008).
The authors thank to the Fundação Calouste Gulbenkian by the financing of the project 96442.
The authors thank to the FTC, Portugal (Strategic Project PEst-C/SAU/UI3282/2013 and UID/NEU/04539/2013), COMPETE-FEDER.
The authors thank Professor Francisco Caramelo from Biophysics Unit - Faculty of Medicine of Coimbra, University of Coimbra, for his support in the calculation of irradiation doses.
Abbreviations
- HCC
Hepatocellular Carcinoma
- p-P53
phosphorylated P53 protein
- ROS
reactive oxygen species
Footnotes
Conflicts of Interest: The authors have no conflicts to disclose.
References
- 1.World Health Organization. GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalende Worldwide in 2012. WHO web site. [Accessed 2011]. http://globocan.iarc.fr/Default.aspx.
- 2.Thomas MB, Zhu AX. Hepatocellular carcinoma: the need for progress. J Clin Oncol. 2005;23:2892–2899. doi: 10.1200/JCO.2005.03.196. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz JM, Ham JM. Treatment of hepatocellular carcinoma. Curr Treat Options Gastroenterol. 2003;6:465–472. doi: 10.1007/s11938-003-0048-z. [DOI] [PubMed] [Google Scholar]
- 4.Gudkov AV, Komarova EA. The role of P53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003;3:117–129. doi: 10.1038/nrc992. [DOI] [PubMed] [Google Scholar]
- 5.Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239–253. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- 6.Katz D, Ito E, Liu FF. On the path to seeking novel radiosensitizers. Int J Radiat Oncol Biol Phys. 2009;73:988–996. doi: 10.1016/j.ijrobp.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Banfi A. Principles of radiotherapy. In: Bonnadona G, Robustelli della Cuna G, editors. Handbook of medical oncology. Milano: Masson; Chicago: Year Book Medical; 1988. pp. 191–196. [Google Scholar]
- 8.Brans B, Linden O, Giammarile F, Tennvall J, Punt C. Clinical applications of newer radionuclide therapies. Eur J Cancer. 2006;42:994–1003. doi: 10.1016/j.ejca.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 9.Keng GH, Sundram FX. Radionuclide therapy of hepatocellular carcinoma. Ann Acad Med Singapore. 2003;32:518–524. [PubMed] [Google Scholar]
- 10.Keng GH, Sundram FX, Yu SW, Somanesan S, Premaraj J, Oon CJ, et al. Preliminary experience in radionuclide therapy of hepatocellular carcinoma using hepatic intra-arterial radio-conjugates. Ann Acad Med Singapore. 2002;31:382–386. [PubMed] [Google Scholar]
- 11.Raoul JL, Boucher E, Rolland Y, Garin E. Treatment of hepatocellular carcinoma with intra-arterial injection of radionuclides. Nature reviews. Nat Rev Gastroenterol Hepatol. 2010;7:41–49. doi: 10.1038/nrgastro.2009.202. [DOI] [PubMed] [Google Scholar]
- 12.Wyszomirska A. Iodine-131 for therapy of thyroid diseases. Physical and biological basis. Nucl Med Rev Cent East Eur. 2012;15:120–123. [PubMed] [Google Scholar]
- 13.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuddihy AR, Bristow RG. The p53 protein family and radiation sensitivity: Yes or no? Cancer Metastasis Rev. 2004;23:237–257. doi: 10.1023/B:CANC.0000031764.81141.e4. [DOI] [PubMed] [Google Scholar]
- 15.Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–221. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 16.Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer. 2004;4:592–603. doi: 10.1038/nrc1412. [DOI] [PubMed] [Google Scholar]
- 17.Kaino M. Alterations in the tumor supressor genes p53, RB, p16/MTS1, and p15/MTS2 in human pancreatic cancer and hepatoma cell lines. J Gastroenterol. 1997;32:40–46. doi: 10.1007/BF01213295. [DOI] [PubMed] [Google Scholar]
- 18.Mamede AC, Pires AS, Abrantes AM, Tavares SD, Gonçalves AC, Casalta-Lopes JE, et al. Cytotoxicity of ascorbic acid in a human colorectal adenocarcinoma cell line (WiDr): in vitro and in vivo studies. Nutr Cancer. 2012;64:1049–1057. doi: 10.1080/01635581.2012.713539. [DOI] [PubMed] [Google Scholar]
- 19.Brito AF, Mendes M, Abrantes AM, Tralhão JG, Botelho MF. Positron emission tomography diagnostic imaging in multidrug-resistant hepatocellular carcinoma: focus on 2-deoxy-2-(18F)Fluoro-D-Glucose. Mol Diagn Ther. 2014;18:495–504. doi: 10.1007/s40291-014-0106-3. [DOI] [PubMed] [Google Scholar]
- 20.Li J, He M, Shen B, Yuan D, Shao C. Alpha particle-induced bystander effect is mediated by ROS via a p53-dependent SCO2 pathway in hepatoma cells. Int J Radiat Biol. 2013;89:1028–1034. doi: 10.3109/09553002.2013.817706. [DOI] [PubMed] [Google Scholar]
- 21.He M, Zhao M, Shen B, Prise KM, Shao C. Radiation-induced intercellular signaling mediated by cytochrome-c via a p53-dependent pathway in hepatoma cells. Oncogene. 2011;30:1947–1955. doi: 10.1038/onc.2010.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang Y, Zhou X, Chen X, Yang G, Wang Q, Rao K, et al. Benzo(a) pyrene-induced mitochondrial dysfunction and cell death in p53-null Hep3B cells. Mutat Res. 2011;726:75–83. doi: 10.1016/j.mrgentox.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Bressac B, Galvin KM, Liang TJ, Isselbacher KJ, Wands JR, Ozturk M. Abnormal structure and expression of p53 gene in human hepatocellular carcinoma. Proc Natl Acad Sci U S A. 1990;87:1973–1977. doi: 10.1073/pnas.87.5.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman SL, Shaulian E, Littlewood T, Resnitzky D, Oren M. Resistance to p53-mediated growth arrest and apoptosis in Hep 3B hepatoma cells. Oncogene. 1997;15:63–70. doi: 10.1038/sj.onc.1201149. [DOI] [PubMed] [Google Scholar]
- 25.Kuo PL. Lin TC, Lin CC. The antiproliferative activity of aloe-emodin is through p53-dependent and p21-dependent apoptotic pathway in human hepatoma cell lines. Life Sci. 2002;71:1879–1892. doi: 10.1016/s0024-3205(02)01900-8. [DOI] [PubMed] [Google Scholar]
- 26.Reiser M, Neumann I, Schmiegel W, Wu PC, Lau JY. Induction of cell proliferation arrest and apoptosis in hepatoma cells through adenoviral-mediated transfer of p53 gene. J Hepatol. 2000;32:771–782. doi: 10.1016/s0168-8278(00)80246-9. [DOI] [PubMed] [Google Scholar]
- 27.Chan KT, Lung ML. Mutant p53 expression enhances drug resistance in a hepatocellular carcinoma cell line. Cancer Chemother Pharmacol. 2004;53:519–526. doi: 10.1007/s00280-004-0767-4. [DOI] [PubMed] [Google Scholar]
- 28.Zhu GN, Zuo L, Zhou Q, Zhang SM, Zhu HQ, Gui SY, et al. Loss of heterozygosity on chromosome 10q22-10q23 and 22q11.2-22q12.1 and p53 gene in primary hepatocellular carcinoma. World J Gastroenterol. 2004;10:1975–1978. doi: 10.3748/wjg.v10.i13.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slebos RJ, Resnick MA, Taylor JA. Inactivation of the p53 tumor suppressor gene via a novel Alu rearrangement. Cancer Res. 1998;58:5333–5336. [PubMed] [Google Scholar]
- 30.Knowles BB, Aden DP. U.S. Patent No. 4, 393,133. Washington: U.S. Patent and Trademark Office; Human hepatoma derived cell line, process for preparation thereof, and uses therefor. 1983
- 31.Brito AF, Abrantes AM, Ribeiro M, Oliveira R, Casalta-Lopes J, Gonçalves AC, et al. Fluorine-18 fluorodeoxyglucose uptake in hepatocellular carcinoma: correlation with Glucose Transporters and p53 expression. J Clin Exp Hepatol. 2015 Jun; doi: 10.1016/j.jceh.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verheij M. Clinical biomarkers and imaging for radiotherapy-induced cell death. Cancer Metastasis Rev. 2008;27:471–480. doi: 10.1007/s10555-008-9131-1. [DOI] [PubMed] [Google Scholar]
- 33.Ross GM. Induction of cell death by radiotherapy. Endocr Relat Cancer. 1999;6:41–44. doi: 10.1677/erc.0.0060041. [DOI] [PubMed] [Google Scholar]
- 34.Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev. 2006;20:1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
- 35.Paillas S, Boudousq V, Piron B, Kersual N, Bardiès M, Chouin N, et al. Apoptosis and p53 are not involved in the anti-tumor efficacy of 125I-labeled monoclonal antibodies targeting the cell membrane. Nucl Med Biol. 2013;40:471–480. doi: 10.1016/j.nucmedbio.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 36.Baumann K. Cell death: multitasking p53 promotes necrosis. Nat Rev Mol Cell Biol. 2012;13:480–481. doi: 10.1038/nrm3401. [DOI] [PubMed] [Google Scholar]
- 37.Andrianantoandro E. p53 Opens the Gates to Necrosis. Sci Signal. 2012;5:ec175. [Google Scholar]
- 38.Riley PA. Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol. 1994;65:27–33. doi: 10.1080/09553009414550041. [DOI] [PubMed] [Google Scholar]
- 39.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 40.Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther. 2008;7:1875–1884. doi: 10.4161/cbt.7.12.7067. [DOI] [PubMed] [Google Scholar]
- 41.Van Pelt WR, Drzyzga M. Beta radiation shielding with lead and plastic: effect on bremsstrahlung radiation when switching the shielding order. Health Phys. 2007;92(2 Suppl):S13–S17. doi: 10.1097/01.HP.0000248127.71398.b1. [DOI] [PubMed] [Google Scholar]