

Summary
To our knowledge, this is the first study using a non-surgical model to provide evidence that malabsorption of endogenous bile acids in a model with an intact small intestine is associated with increased colon neoplasia. This finding has potentially important clinical implications.
Abstract
Although epidemiological evidence in humans and bile acid feeding studies in rodents implicate bile acids as tumor promoters, the role of endogenous bile acids in colon carcinogenesis remains unclear. In this study, we exploited mice deficient in the ileal apical sodium-dependent bile acid transporter (ASBT, encoded by SLC10A2) in whom fecal bile acid excretion is augmented more than 10-fold. Wild-type and Asbt-deficient (Slc10a2 −/−) male mice were treated with azoxymethane (AOM) alone to examine the development of aberrant crypt foci, the earliest histological marker of colon neoplasia and a combination of AOM and dextran sulfate sodium to induce colon tumor formation. Asbt-deficient mice exhibited a 54% increase in aberrant crypt foci, and 70 and 59% increases in colon tumor number and size, respectively. Compared to littermate controls, Asbt-deficient mice had a striking, 2-fold increase in the number of colon adenocarcinomas. Consistent with previous studies demonstrating a role for muscarinic and epidermal growth factor receptor signaling in bile acid-induced colon neoplasia, increasing bile acid malabsorption was associated with M3 muscarinic and epidermal growth factor receptor expression, and activation of extracellular signal-related kinase, a key post-receptor signaling molecule.
Introduction
Primary bile acids are synthesized in the liver from cholesterol, N-acyl amidated to taurine or glycine, secreted into bile and, in most mammals, stored in the gallbladder before meal-stimulated release into the proximal small intestine where they facilitate lipid digestion. Although small quantities of bile acids are reabsorbed passively throughout the gut, ~95% of intestinal bile acid uptake is mediated in the distal small intestine by the ileal apical sodium-dependent bile acid transporter (ASBT, encoded by SLC10A2) (1). Bile acids absorbed from the gut return to the liver via the portal venous circulation and are re-excreted into bile and the small bowel. This enterohepatic circulation minimizes bile acid loss in stool. ASBT-deficiency markedly impairs bile acid absorption and fecal bile acid excretion is greatly augmented in Asbt-deficient (Slc10a2 −/−) compared to wild-type (WT) male mice (2).
Although chronic or intermittent diarrhea is the most overt clinical manifestation of bile acid malabsorption, the long-term consequences of persistently exposing colon epithelial cells to increased bile acid levels are uncertain. One concern is that by direct or indirect actions on colon epithelial cells fecal bile acids promote colon neoplasia. Whereas colon cancer arises from progressive mucosal dysplasia associated with somatic gene mutations, environmental factors can accelerate and augment this process. Diets rich in meat and fat increase both fecal bile acid levels and colon cancer risk (3).
Surprisingly little is known about ASBT expression and activity in colon cancer. Wang et al. (4). reported a greater than 2-fold increase in the risk of colon adenomas in persons with SLC10A2 gene polymorphisms, but a case–control study found no increase in colon cancer risk with common SLC10A2 gene variants (5). These studies were limited by small sample sizes, failure to control for diet, drugs, cholecystectomy and other factors that modulate intestinal bile acid levels and composition, and did not correlate SLC10A2 gene polymorphisms with either tumor size or progression. As growth factors, bile acids are more likely to act as tumor promoters than initiators (6–8).
In rodent models of chemical carcinogen-induced colon neoplasia, instillation of exogenous bile acids into the gut promotes epithelial dysplasia and tumor formation (3). Even without a genetic predisposition to colon neoplasia or exposure to a carcinogen, mice fed a human secondary bile acid, deoxycholic acid, for 8–10 months developed colon cancer (8). Collectively, these observations provide evidence that excess levels of exogenous bile acids can promote colon neoplasia. In contrast, the effects of endogenous bile acids on the development of chemical carcinogen-induced colon neoplasia in rodents have only been examined in surgical models where large portions of the small bowel were resected or bypassed to increase bile acid flux into the colon. An important limitation of those studies is that bowel resection disrupts normal intestinal function and results in malabsorption of many luminal constituents in addition to bile acids. Indeed, studies using that approach found increased chemical carcinogen-induced colon neoplasia in rats following resection of either the proximal small intestine or the distal small intestine (9,10). To overcome the limitations of surgical approaches we used Asbt-deficient mice as an in vivo model of bile acid malabsorption and to assess the cancer-promoting potential of chronically exposing colon epithelial cells to elevated levels of endogenous bile acids. In Asbt-deficient mice, the lack of bile acid uptake in the distal small intestine, which we recently documented using novel in vivo imaging (11,12), results in more than 10-fold increased fecal bile acids (2). Unlike surgical models, fecal weight is not increased in Asbt-deficient mice despite a significant decrease in colon transit time (2).
Materials and methods
Animals
We used male mice with targeted disruption of the Asbt gene (Slc10a2 −/− mice) and wild-type (WT) male mice, all backcrossed more than 10 generations on a C57BL/6J genetic background (2). For all experiments, mice were housed under identical conditions in a pathogen-free environment with a 12:12-h light/dark cycle and free access to water and standard mouse chow (the Teklad Global 18% Protein Extruded Rodent Diet contains as percentage of calories: 24% protein, 18% fat and 58% fiber). All animal studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals prepared by the U.S. National Academy of Sciences. Studies conducted at Wake Forest University School of Medicine were approved by that school’s Institutional Animal Care and Use Committee. Animal studies conducted in Baltimore were approved by both the Institutional Animal Care and Use Committee at the University of Maryland School of Medicine and the Research and Development Committee at the VA Maryland Health Care System.
Aberrant crypt foci study
Asbt-null (Slc10a2 −/−; N = 10 mice) and WT mice (N = 11 mice) received 7.5mg/kg azoxymethane (AOM) by intraperitoneal injection weekly for 10 weeks beginning at 4 weeks of age. Mice were weighed weekly and none exhibited weight loss during or after AOM dosing; they were killed 20 weeks after the first AOM injection (24 weeks of age). The small intestine and colon were isolated from each mouse. The small intestine was divided into four equal segments, slit longitudinally and rinsed with ice-cold phosphate-buffered saline. Segments were laid flat in a container with filter paper soaking in phosphate-buffered formalin. All segments were fixed overnight in phosphate-buffered formalin. Similarly, colons were cleaned and fixed in formalin. After fixation, tissues were rinsed and stored in 60% ethanol. In WT and Slc10a2 −/−mice, no macroscopic lesions were present in either the small intestine or colon. Colons were stained in 0.2% methylene blue in phosphate-buffered saline for 1–2min, rinsed briefly in phosphate-buffered saline, and examined at 40× magnification for aberrant crypt foci (ACF).
Tumor study
Slc10a2 −/− (N = 15 mice) and WT mice (N = 20 mice), received 7.5mg/kg body weight AOM by intraperitoneal injection weekly for 4 weeks beginning at 13–21 weeks of age (times of AOM initiation were matched between Slc10a2 −/− and WT mice). After the last AOM injection, the animals’ drinking water was supplemented with 2.5% dextran sulfate sodium (DSS) for 5 days. Three Slc10a2 −/− mice died within 1 week after DSS treatment and were not analyzed. Control mice (three Slc10a2 −/− and four WT mice) received neither AOM nor DSS. Treated and control mice were killed 20 weeks after the first AOM injection; after harvest, colon length was measured, and colons were opened longitudinally, photographed (Nikon SMZ1500 dissecting microscope) and inspected visually by an investigator masked to genotype and treatment who measured tumor number and size. Tumor volume was calculated using the equation, volume = 1/2 (length × width2) (6). Sections of normal colon and tumors were snap frozen to measure protein, stored in RNAlater (Sigma–Aldrich, St Louis, MO) to determine mRNA expression, and the remaining tissue was fixed in 4% paraformaldehyde and paraffin-embedded; 5 µm sections were used for hematoxylin and eosin and immunostaining. A senior pathologist masked to mouse genotype or treatment classified colon tumors in hematoxylin and eosin-stained sections as adenomas or adenocarcinomas according to consensus recommendations (13).
Quantitative real-time polymerase chain reaction
To examine the expression of intestinal transcripts, total RNA from tumors and adjacent normal colon was extracted from tissue stored in RNAlater. Five micrograms of RNA were used for first-strand complementary DNA synthesis using the Superscript III First Strand Synthesis System for RT-PCR (Invitrogen). Quantitative real-time polymerase chain reaction was performed with 50ng complementary DNA, the SYBR Green PCR Master Mix (Applied Biosystems) and specific primers at a final concentration of 0.5 μM in sample volumes of 20 μl. The primer sequences shown in Table 1 were designed to span introns (National Center for Biotechnology Information nucleotide database SIM-4 gene alignment program and online software; http://www.genscript.com/index.html). qPCR was performed using the 7900HT Fast System and SYBR Green Master Mix (ABI) following the manufacturer’s protocol. PCR conditions were 5min at 95°C, followed by 37 cycles of 95°C for 15 s, 60°C for 20 s and 72°C for 40 s and a final cycle at 95°C for 15 s. Data were analyzed using ABI instrument software (SDS 2.1). Expression of GAPDH was used as an endogenous control for normalization. All samples were run in triplicate and the C T (2–ΔΔCT) method was used to compare samples when assessing the relative expression of transcripts.
Table 1.
qPCR primer sequences used
| Protein | Gene | Forward | Reverse |
|---|---|---|---|
| FXR | Nr1h4 | TCCACAACCAAGTTTTGCAG | TCTCTGTTTGTTGTACGAATCCA |
| MRP3 | Abcc3 | AGAGCTGGGCTCCAAGTTCT | TGGTGTCTCAGGTAAAACAGGTAGCA |
| Ostβ | Slc51b | GTATTTTCGTGCAGAAGATGCG | TTTCTGTTTGCCAGGATGCTC |
| Ostα | Slc51a | TACAAGAACACCCTTTGCCC | CGAGGAATCCAGAGACCAAA |
| Cox2 | Ptgs2 | AGCCTTCTCCAACCTCTCCT | CAGGGATGAACTCTCTCCGT |
| cMyc | cMyc | CAGCAGCGACTCTGAAGAAG | GACTCCGACCTCTTGGGA |
| β-catenin | Ctnnb1 | CCTGAGGGTACCTGAAGCTC | GCTTGAGTAGCCATTGTCCA |
| Cyclin D1 | CycD1 | CAACGCACTTTCTTTCCAGA | GACTCCAGAAGGGCTTCAAT |
| Matrix metalloproteinase7 | Mmp7 | CTGTTCCCGGTACTGTGATG | TCACAGCGTGTTCCTCTTTC |
| M3R | Chrm3 | TGGTGTGTTCTTCCTTGGAC | ACCCAGGAAGAGCTGATGTT |
| EGFR | Egfr | CCACGCCAACTGTACCTATG | CACAATCCCAGTGGCAATAG |
Immunohistochemical staining
To identify and quantify protein expression, formalin-fixed paraffin-embedded tissue sections were immunostained with specific antibodies against M3 muscarinic receptors (M3R) (Alomone Labs, Jerusalem, Israel), extracellular signal-related kinase (ERK) and phospho-ERK (Cell Signaling Technology), and epidermal growth factor receptors (Sigma–Aldrich). Tumor sections were examined with a Nikon 80i photomicroscope at 200× magnification. Sections were first reviewed and scored by a senior pathologist (C.D.) masked to tissue origin, and immunostaining was then quantified using Image-Pro Plus software (version 5.1; Media Cybernetics, Silver Spring, MD). To minimize variation, all tissue sections were examined and photographed using the same microscope settings.
Ki67 staining
To examine cell proliferation, Ki67 staining was performed as described by Holt et al. (14). Only the distal half of the colon and complete crypts were analyzed by investigators masked to genotype and treatment. To measure changes in the distribution of Ki67 staining, crypts were divided equally into five zones, starting at the base of the crypt (zone 1) and extending to the luminal surface (zone 5). The ‘labeling index’ was calculated as the percentage of cells in a crypt zone that stained for Ki67 (14); a minimum of 1000 colonocytes was evaluated per mouse colon.
Statistics
Data are presented as average value ± standard error of the mean. The unpaired Student’s t test (assuming unequal variance) was used for comparing continuous variables between two independent groups. A logarithm transformation (Log(tumor volume + 1)) was applied to tumor volume so the transformed variable approximately followed a normal distribution. Then, because multiple tumors could be present in one mouse, a linear mixed model for clustered data was applied to compare the transformed tumor volume between WT and Asbt-deficient mice. Two-way analysis of variance with one between-subject factor (WT versus Asbt-deficient) and one within-subject factor (normal tissue versus tumor tissue) was applied to compare gene expression, followed by post hoc tests with Tukey–Kramer’s adjustment for P values. Fisher’s exact test was used for comparing proportions. Differences were considered significant when P < 0.05.
Results
ACF provide the earliest histological evidence of colon neoplasia, sometimes appearing in murine models within a few weeks of carcinogen treatment (13). To investigate the effects of Asbt deficiency on colon neoplasia, we examined ACF formation in WT and Slc10a2 −/− mice treated with AOM, a commonly used experimental carcinogen (6). Starting at age of 4 weeks, 11 WT and 10 Slc10a2 −/− mice received 7.5mg/kg AOM by intraperitoneal injection once each week for 10 weeks. Mice were killed 20 weeks after the first AOM injection and their colons were harvested, stained with methylene blue and examined for ACF.
ACF were identified microscopically in methylene blue-stained harvested colon tissue by readily identifiable features including enlarged crypts, a thickened layer of methylene blue-stained epithelial cells, slit-shaped luminal openings, increased pericryptal space and elevation above the focal plane (Figure 1A, inset) (13). We found a statistically significant, 60% increase in the number of ACF in colons from AOM-treated Slc10a2 −/− mice compared to those from WT mice (P = 0.03; Figure 1A). Based on this promising proof-of-concept that increased levels of fecal bile acids promote colon neoplasia in Asbt-deficient mice, we examined the effects of Asbt deficiency on more advanced colon neoplasia.
Figure 1.

Asbt deficiency promotes colon neoplasia. (A) Colons were harvested from 11 WT and 10 Slc10a2 −/− AOM-treated mice killed 20 weeks after starting AOM injections (7.5mg/kg). Excised colons were stained with methylene blue and examined for ACF. Significantly more ACF were detected in colons from Slc10a2 −/− compared to WT mice (P = 0.03, Student’s unpaired t-test). Error bars denote SEM. Inset: Representative microscopic images of ACF stained with methylene blue. Original magnification, ×40. (B) Experimental design and mouse weights over the course of the 20-week study. Vertical arrows on the horizontal axis indicate intraperitoneal injection with 7.5mg/kg AOM at days 1, 8, 15 and 22. Vertical blue band indicates that 2.5% DSS was added to the drinking water of AOM-treated mice on days 22 through 26. Twenty WT and 15 Slc10a2 −/− mice were treated with AOM plus DSS; three Slc10a2 −/− mice died immediately after DSS treatment. Four WT and three Slc10a2 −/− mice were treated with vehicle alone. Error bars denote SEM. (C) Numbers of colon tumors in WT and Slc10a2 −/− mice treated with AOM plus DSS. Horizontal bars indicate mean numbers of colon tumors, 2.3 and 3.9 tumors per WT and Slc10a2 −/− mouse colon, respectively (P = 0.02). No tumors were observed in colons from WT and Slc10a2 −/− mice treated with vehicle alone. (D) Total tumor volume was calculated in colons from WT and Slc10a2 −/− mice treated with AOM plus DSS. A linear mixed model for repeated measures was applied to compare the logarithm transformed tumor volume between WT mice and ASBT mice. A significant difference between the two groups was found (mean difference in log scale = 0.54, P = 0.04). Error bars denote SEM.
Even though they may develop ACF, some mouse strains, like C57BL/6, resist progression to colon tumor formation following treatment with AOM alone (3), a limitation circumvented by adding DSS to their drinking water. We treated mice using the study design illustrated in Figure 1B; 20 WT and 15 Slc10a2 −/− male littermates were treated with AOM (7.5mg/kg by intraperitoneal injection once each week for 4 weeks) plus 2.5% DSS (added to the drinking water for 5 days). Four WT and three Slc10a2 −/− mice treated with vehicle alone served as additional controls. As shown in Figure 1B, Asbt-deficient mice weighed approximately 10% less than WT littermates; this significant difference (P < 0.05) persisted throughout the study period, including a transient drop in body weight immediately after DSS treatment; three Slc10a2 −/− mice died within a week of DSS treatment. The small differences in body weight between male Slc10a2 −/− and WT mice, reported previously (2), did not impact colon length (data not shown).
Although no colon tumors were detected in vehicle-treated mice, most WT and Asbt-deficient animals developed tumors (Figure 1C). As shown in Figure 1C and D, Asbt deficiency was associated with a 70% increase in the mean number of colon tumors per mouse (P = 0.02) and 59% increase in mean tumor size (P = 0.04). Asbt deficiency also significantly increased the multiplicity of colon tumors; nine WT mice versus only one Asbt-deficient mouse had fewer than two tumors (P = 0.049, Fisher’s two-tailed exact test; Figure 1C).
Increased cell proliferation is a hallmark of neoplastic transformation and bile acids stimulate colonocyte proliferation in vitro (15). To examine the actions of increased fecal bile acids on colonocyte proliferation in vivo, we measured Ki67 immunostaining (Figure 2A). As shown in Figure 2B, Asbt deficiency differentially affected Ki67 immunostaining depending upon the crypt zone examined; in colons from Slc10a2 −/− compared to those from WT mice there were significantly fewer Ki67-stained cells at the crypt base (zone 1); the reverse was observed at the crypt apex (zone 5) (both P < 0.05). The shift of proliferating cells to the outer portions of the crypt in Slc10a2 −/− mice likely represents an early manifestation of dysplasia. We measured colonocyte apoptosis using caspase-3 immunostaining; as we observed previously (6) caspase-3-stained cells were an order of magnitude fewer than Ki67-stained cells and there were no significant differences in either normal epithelial or tumor cell apoptosis when comparing WT and Asbt-deficient mice (data not shown). These findings support the concept that fecal bile acids act primarily as growth factors that promote cell proliferation.
Figure 2.
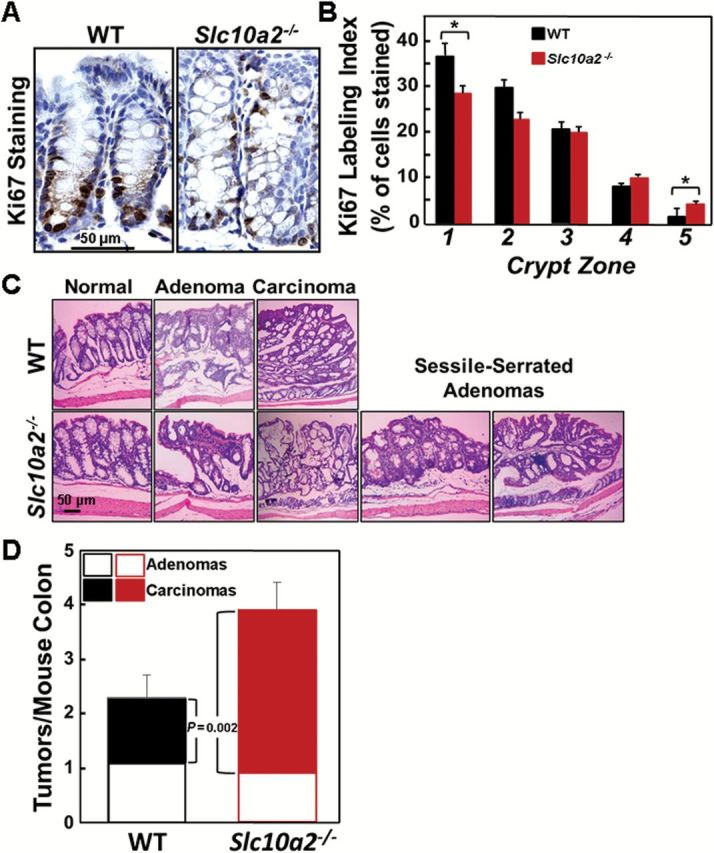
Asbt deficiency promotes colonocyte proliferation and advanced colon neoplasia. (A) Representative images of Ki67 staining in colonic crypts from WT and Slc10a2 −/− mice. (B) Effect of Asbt deficiency on proliferation of normal colonocytes. The Ki67 labeling index was measured in normal crypts from the distal half of the colon. Crypts were divided into five equal zones, starting from the base (zone 1) and extending to the luminal surface (zone 5), and a minimum of 1000 crypt colonocytes was assessed in the colon from each mouse (N = 20 WT and 12 Slc10a2 −/− mice). Note increased Ki67 staining in upper portions of crypts in normal colons from Asbt-deficient mice. (*P = 0.05 compared to crypts from WT mice). (C) Representative hematoxylin and eosin-stained sections of normal colon, adenomas, adenocarcinomas and sessile-serrated adenomas from WT and Slc10a2 −/− mice. Sessile-serrated adenomas were observed only in colons from AOM/DSS-treated Slc10a2 −/− mice. (D) Asbt deficiency promotes progression from adenoma to adenocarcinoma. Bar graph shows distribution of colon adenomas and adenocarcinomas in WT and Slc10a2 −/− mice treated with AOM plus DSS. There was no difference in the numbers of adenomas between the two groups but the number of adenocarcinomas was significantly greater in Slc10a2 −/− compared to WT mice (P = 0.002). Error bars denote SEM.
Mouse colon tumors represented both adenomas and adenocarcinomas; two Asbt-deficient mice, but no WT mice, developed sessile-serrated adenomas (Figure 2C), a biomarker for activation of epidermal growth factor receptor signaling (16). Notably, as shown in Figure 2D, the increased tumor burden in Asbt-deficient mice resulted from more adenocarcinomas, not adenomas. Whereas the overall number of adenomas was unchanged, Asbt-deficient mice had a nearly 2-fold increase in the number of adenocarcinomas (P = 0.002).
In the colon, two bile acid transporters expressed on the colonocyte basolateral membrane mediate export of passively absorbed bile acids into the enterohepatic circulation; heteromeric Organic Solute Transporter (OSTα-OSTβ) and multidrug resistance-associated protein-3 (MRP3) (17). Bile acid induced changes in OSTα-OSTβ expression are regulated in part by the bile acid nuclear receptor farnesoid X receptor (FXR) (17). To explore the consequences of increased fecal bile acids, we compared the expression of genes for FXR and these bile acid transporters in tissue from Asbt-deficient to that from WT mice.
In humans and murine colon neoplasia models, expression of the nuclear bile acid receptor FXR is reduced in colon carcinoma compared to adjacent normal mucosa (18). Thus, it was not surprising to find that in adenocarcinomas from both WT and Asbt-deficient mice Fxr expression was significantly reduced compared to normal colon (Figure 3A). Similar changes were observed for the genes encoding Mrp3 (Abcc3), Ostα and Ostβ (Figure 3A), most likely reflecting regulation of bile acid transporter expression by Fxr.
Figure 3.

Increased Mmp7 gene expression, and muscarinic and EGF receptor expression, and activation of post-receptor signaling in tumors from Slc10a2 −/− mice. (A) qRT-PCR of colon epithelial cell bile acid nuclear receptor and transporters. Expression of genes for Fxr, Mrp3 (Abcc3) and Ostβ was significantly reduced in adenocarcinomas from WT and Slc10a2 −/− mice compared to adjacent normal colon (*P < 0.05, **P < 0.01, ***P < 0.001; n = 9–11). (B) qRT-PCR of markers for colon cancer progression. Expression of Ptgs2, cMyc, CycD1 and Mmp7 was increased in adenocarcinomas from WT and Slc10a2 −/− mice compared to adjacent normal colon (*P < 0.05, **P < 0.01; n = 9–11). Mmp7 expression was significantly increased in normal colon from Slc10a2 −/− mice compared to normal colon from WT mice (**P < 0.01; ***P < 0.001). (C) Representative images of M3R staining in normal colon tissue and adenocarcinomas from WT and Slc10a2 −/− mice. (D) M3R staining is increased in tumors compared to normal colon epithelium from both WT and Asbt-deficient mice (left panel). Nl, normal tissue. Bars represent mean ± SEM for staining intensity score measured in 0.5 increments by a senior pathologist on a scale from 0, no staining to 3, maximal staining (**P < 0.01; n = 10–13). M3R staining was significantly increased in tumors from Asbt-deficient (KO) mice compared to those from WT mice (right panel). Bars represent mean ± SEM for staining intensity expressed in arbitrary units (*P < 0.05; n = 10–13).
To gain additional insight into the mechanisms underlying the promotion of colon neoplasia in Asbt-deficient mice, we examined the relative expression of genes commonly associated with colon cancer progression. As shown in Figure 3B, expression levels of the genes for cyclooxygenase 2 (Ptgs2), cMyc and cyclinD1 (CycD1) in both WT and Asbt-deficient mice were significantly increased in adenocarcinomas. However, there was no significant difference in the relative expression of these genes in normal colon and tumors from Asbt-deficient mice when compared with those from WT mice (solid black and red bars in Figure 3B). This suggests that altered expression of Ptgs2, cMyc, Ctnnb1 and CycD1 is most probably a consequence of neoplastic transformation independent of Asbt deficiency.
Consistent with its important role in colon cancer progression (7), expression of the gene for matrix metalloproteinase-7 (Mmp7) was strikingly induced in murine colon tumors; 40- to 400-fold in tumors versus adjacent normal colon from WT mice and Asbt-deficient mice (P < 0.001; Figure 3B). Moreover, in normal tissue from Asbt-deficient compared to WT mice, Mmp7 expression was increased 8-fold (P < 0.01; Figure 3B), suggesting that induction of Mmp7 expression in normal colon may be relevant to bile acid-induced promotion of colon neoplasia.
Muscarinic receptors and ligands play important roles in colon neoplasia (6,7); M3R are over-expressed in colon cancer and their activation stimulates colon cancer cell proliferation. In animal models of genetic and sporadic colon cancer, M3R knockout or inactivation attenuates murine colon epithelial cell proliferation and neoplasia (6,19). We used immunohistochemistry to examine M3R expression in normal colon epithelium and carcinomas from WT and Asbt-deficient mice. As illustrated in Figure 3C and D, left panel, in both WT and Asbt-deficient mice we observed significantly increased M3R staining in colon cancers compared to normal tissue and a more modest, but statistically significant, increase in M3R expression in tumors from Asbt-null compared to WT mice (Figure 3D, right panel; P < 0.05). In tumors from Asbt-deficient animals, we detected nearly 2-fold increased expression of Chrm3, the gene encoding M3R (Figure 4A), a finding indicative of M3R activation (20). Chrm3 over-expression in Asbt-deficient mice was of similar magnitude to that observed when AOM-treated WT mice were fed bethanachol, a muscarinic receptor agonist (21); bethanachol treatment had also stimulated 30 and 52% increases in tumor number and size (21), of similar magnitude to what we observed in Asbt-deficient mice (Figure 1C and D). Moreover, like Asbt deficiency (Figure 2D), bethanachol treatment increased the numbers of colon adenocarcinomas, not adenomas (21).
Figure 4.
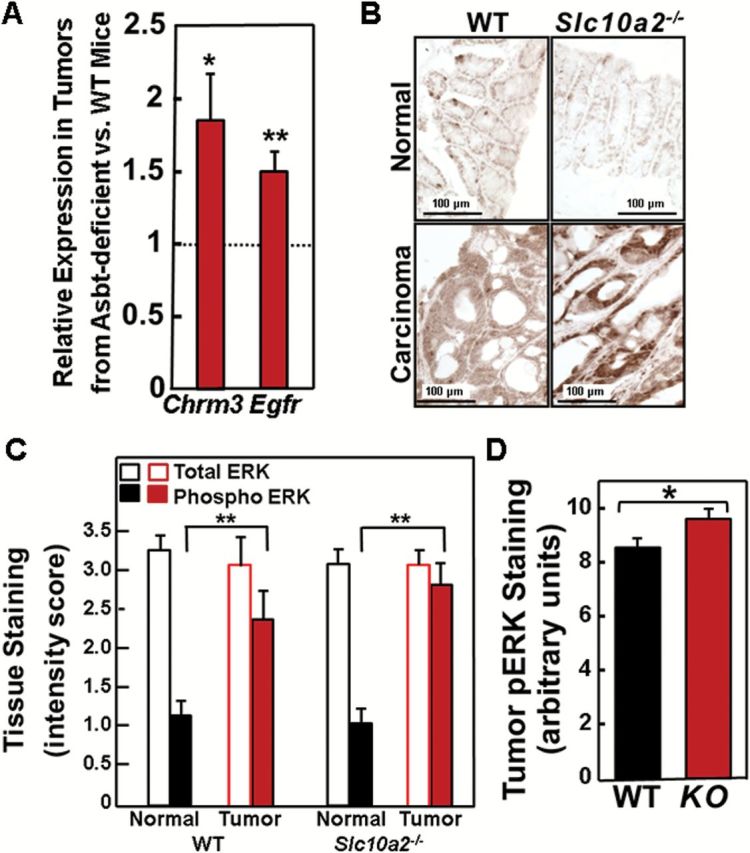
Increased muscarinic and EGF receptor expression, and activation of post-receptor signaling in tumors from Slc10a2 −/− mice. (A) qRT-PCR of genes for type 3 muscarinic (Chrm3) and epidermal growth factor (Egfr) receptors. Expression of Chrm3 and Egfr was significantly increased in tumors from Slc10a2 −/− mice compared to those from WT mice (*P < 0.05, **P < 0.01; n = 9–11). (B) Representative images of phospho-ERK staining in normal colon tissue and adenocarcinomas from WT and Slc10a2 −/− mice. (C) Total ERK staining is unchanged but phospho-ERK staining is significantly increased in tumors compared to normal colon epithelium from WT and Asbt-deficient mice. Bars represent mean ± SEM for staining intensity score measured in 0.5 increments by a senior pathologist on a scale from 0, no staining to 3, maximal staining (**P < 0.01; n = 10–13). (D) Phospho-ERK (pERK) staining was significantly increased in tumors from Asbt-deficient (KO) mice compared to those from WT mice. Bars represent mean ± SEM for staining intensity expressed in arbitrary units (*P < 0.05; n = 10–13).
In seeking evidence of post-M3R signal transduction, we detected overexpression of mRNA for epidermal growth factor receptors in tumors from Asbt-deficient mice (Figure 4A), and in both WT and Asbt-deficient mice, immunostaining for epidermal growth factor receptor protein was increased in tumors compared to normal tissue (immunohistochemical staining scores: 2.88±0.18 versus 2.04±0.17 for WT mice; 3.05±0.09 versus 1.90±0.16 for Asbt-deficient mice; both P < 0.005) (Figure 4B). Whereas normal tissue and tumors from WT and Asbt-deficient animals had similar levels of total ERK, in both genotypes levels of phospho-ERK were significantly increased in tumors compared to normal tissue (Figure 4B and C). Notably, in tumors from Asbt-deficient mice we detected a modest but statistically significant increase in ERK phosphorylation (P < 0.05; Figure 4D), supporting increased ERK activation.
Discussion
In the current study, we used mice deficient in the predominant intestinal bile acid transporter, ASBT, to show that increased spillage of endogenous bile acids into the feces augments the number of ACF and tumors in the colon, and increases tumor size. Notably, in Asbt-deficient mice, the number of adenocarcinomas, not adenomas, was increased. This observation of more advanced neoplasia in Asbt-deficient mice supports the concept that increased fecal bile acids stimulate tumor promotion, not initiation. This concept also agrees with the natural history of the Asbt-deficient mouse; over the period 2002–2013 we maintained 522 Asbt-deficient mice (295 on a C57BL/6 genetic background) on a standard rodent chow diet for 6 months or longer without observing any increased tumor-related mortality. Thus, unlike the situation in mice fed human bile acids for 8–10 months (8) or surgical intestinal resection or bypass (9,10), our findings reveal that impaired intestinal transport of endogenous bile acids is sufficient to promote colon neoplasia.
Previously, using human colon cancer cells, we showed that bile acids activate M3Rs and stimulate cross-talk with epidermal growth factor receptors by a mechanism involving activation of MMP7 (22); muscarinic receptor activation robustly induces MMP7 mRNA expression (7,21). In fact, in Asbt-deficient mice we detected robust overexpression of MMP7 mRNA in both normal colon tissue as well as colon tumors. Since crosstalk between M3 muscarinic and epidermal growth factor receptors is a prominent feature of proliferative signaling in human colon cancer cells (15), our finding of activated ERK signaling in Asbt-deficient mouse colon provides additional support for a mechanistic interaction between M3 muscarinic and epidermal growth factor receptors in mediating the actions of increased fecal bile acids (15). Similar outcomes in mice that ingested bethanachol, a muscarinic receptor agonist, and Asbt-deficient mice with increased spillage of bile acids into the colon lends additional support to this proposed mechanism. Collectively, these observations support bile acid-induced activation of muscarinic receptor signaling as a mechanism whereby increased fecal bile acids induce Mmp7 expression and promote colon neoplasia in Asbt-deficient mice.
To our knowledge, this is the first study using a non-surgical model to suggest that endogenous bile acids can promote colon neoplasia in vivo. Although bile acids such as deoxycholate were shown to induce formation of reactive oxygen and nitrogen species and oxidative DNA damage, and can stimulate muscarinic receptor signaling in colon-derived cell lines, we recognize that evidence in the present study is insufficient to prove that bile acids were directly responsible for the increased neoplasia in vivo. Genetic ablation of Asbt is much more selective than intestinal resection, but bile acid malabsorption is likely to also affect the absorption of other small molecules. As such, the concentration of other endogenous and dietary metabolites may be altered in the soluble phase of the colonic contents. It is also noteworthy that changes in bile acid spectrum and concentration can impact the composition of the gut microbiota and vice versa. Since there is emerging evidence that changes in gut microbial metabolism can affect the development of colon cancer in genetically susceptible models or after treatment with tumor initiators such as AOM/DSS, it is also possible that bile acid malabsorption-associated changes in the gut microbiota may play a role in development of colon neoplasia (23–25).
Despite these limitations, our findings have potentially important clinical implications. Recent studies suggest that idiopathic bile acid malabsorption is not uncommon in the general population and may be a major cause of chronic diarrhea and diarrhea-predominant irritable bowel syndrome. Bile acid malabsorption is also common in inflammatory bowel disease involving the terminal ileum and bariatric surgical procedures such as Roux-en-Y gastric bypass which may increase bile acid flux through the colon. Bile acid diarrhea is a side effect of chemotherapeutic agents such as lenalidomide, and may be an underappreciated effect of several classes of FDA-approved drugs which inhibit bile acid transport by ASBT in vitro (26). Finally, drugs that specifically inhibit ASBT are being developed to treat hypercholesterolemia and constipation; chronic use of such agents may unintentionally promote colon neoplasia.
Supplementary Material
Supplementary Figure 1 can be found at http://carin.oxfordjournals.org/
Funding
National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (DK093406, DK067872 to J.P.R., DK047987 to P.A.D.); VA Merit award (1BX002129 to J.P.R.).
Supplementary Material
Acknowledgements
We thank Dr Rena Lapidus, Stewart and Marlene Greenebaum Cancer Center, University of Maryland School of Medicine for help with AOM/DSS treatment.
Conflict of Interest Statement: PAD is listed as the inventor on two patents owned by Wake Forest University related to screening technology for ASBT inhibitors. PAD has previously served as a consultant for Lumena Pharmaceutical and GlaxoSmithKline. The other authors report no conflicts.
Glossary
Abbreviations
- ACF
aberrant crypt foci
- AOM
azoxymethane
- ASBT
apical sodium-dependent bile acid transporter
- DSS
dextran sulfate sodium
- ERK
extracellular signal-related kinase
- FXR
farnesoid X receptor
- M3R
M3 muscarinic receptors
- MMP7
matrix metalloproteinase-7
- WT
wild type
References
- 1. Dawson P. (2012) Bile formation and the enterohepatic circulation. In Johnson L.R. (ed) Physiology of the Gastrointestinal Tract. Academic Press, Oxford, pp. 1461–1484. [Google Scholar]
- 2. Dawson P.A., et al. (2003) Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J. Biol. Chem., 278, 33920–33927. [DOI] [PubMed] [Google Scholar]
- 3. Flynn C., et al. (2007) Deoxycholic acid promotes the growth of colonic aberrant crypt foci. Mol. Carcinog., 46, 60–70. [DOI] [PubMed] [Google Scholar]
- 4. Wang W., et al. (2001) An association between genetic polymorphisms in the ileal sodium-dependent bile acid transporter gene and the risk of colorectal adenomas. Cancer Epidemiol. Biomarkers Prev., 10, 931–936. [PubMed] [Google Scholar]
- 5. Grünhage F., et al. (2008) Effects of common haplotypes of the ileal sodium dependent bile acid transporter gene on the development of sporadic and familial colorectal cancer: a case control study. BMC Med. Genet., 9, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raufman J.P., et al. (2008) Genetic ablation of M3 muscarinic receptors attenuates murine colon epithelial cell proliferation and neoplasia. Cancer Res., 68, 3573–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xie G., et al. (2009) Acetylcholine-induced activation of M3 muscarinic receptors stimulates robust matrix metalloproteinase gene expression in human colon cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol., 296, G755–G763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bernstein C., et al. (2011) Carcinogenicity of deoxycholate, a secondary bile acid. Arch. Toxicol., 85, 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scudamore C.H., et al. (1983) Effects of small bowel transection, resection, or bypass in 1,2-dimethylhydrazine-induced rat intestinal neoplasia. Gastroenterology, 84, 725–731. [PubMed] [Google Scholar]
- 10. Williamson R.C., et al. (1978) Promotion of azoxymethane-induced colonic neoplasia by resection of the proximal small bowel. Cancer Res., 38, 3212–3217. [PubMed] [Google Scholar]
- 11. Vivian D., et al. (2014) Design and evaluation of a novel trifluorinated imaging agent for assessment of bile acid transport using fluorine magnetic resonance imaging. J. Pharm. Sci., 103, 3782–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vivian D., et al. (2014) In vivo performance of a novel fluorinated magnetic resonance imaging agent for functional analysis of bile acid transport. Mol. Pharm., 11, 1575–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boivin G.P., et al. (2003) Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology, 124, 762–777. [DOI] [PubMed] [Google Scholar]
- 14. Holt P.R., et al. (1997) Is Ki-67 a better proliferative marker in the colon than proliferating cell nuclear antigen? Cancer Epidemiol. Biomarkers Prev., 6, 131–135. [PubMed] [Google Scholar]
- 15. Cheng K., et al. (2005) Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem. Pharmacol., 70, 1035–1047. [DOI] [PubMed] [Google Scholar]
- 16. Bongers G., et al. (2012) A role for the epidermal growth factor receptor signaling in development of intestinal serrated polyps in mice and humans. Gastroenterology, 143, 730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dawson P.A. (2011) Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb. Exp. Pharmacol., 201, 169–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Gottardi A., et al. (2004) The bile acid nuclear receptor FXR and the bile acid binding protein IBABP are differently expressed in colon cancer. Dig. Dis. Sci., 49, 982–989. [DOI] [PubMed] [Google Scholar]
- 19. Raufman J.P., et al. (2011) Muscarinic receptor subtype-3 gene ablation and scopolamine butylbromide treatment attenuate small intestinal neoplasia in Apcmin/+ mice. Carcinogenesis, 32, 1396–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song P., et al. (2008) Activated cholinergic signaling provides a target in squamous cell lung carcinoma. Cancer Res., 68, 4693–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peng Z., et al. (2013) Cholinergic muscarinic receptor activation augments murine intestinal epithelial cell proliferation and tumorigenesis. BMC Cancer, 13, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheng K., et al. (2007) Matrix metalloproteinase-7-catalyzed release of HB-EGF mediates deoxycholyltaurine-induced proliferation of a human colon cancer cell line. Biochem. Pharmacol., 73, 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Belcheva A., et al. (2014) Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell, 158, 288–299. [DOI] [PubMed] [Google Scholar]
- 24. Donohoe D.R., et al. (2014) A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov., 4, 1387–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Louis P., et al. (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol., 12, 661–672. [DOI] [PubMed] [Google Scholar]
- 26. Zheng X., et al. (2009) Computational models for drug inhibition of the human apical sodium-dependent bile acid transporter. Mol. Pharm., 6, 1591–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.