

Abstract
C5′-Hydrogen atoms are frequently abstracted during DNA oxidation. The oxidized abasic lesion 5′-(2-phosphoryl-1,4-dioxobutane) (DOB) is an electrophilic product of the C5′-radical. DOB is a potent irreversible inhibitor of DNA polymerase β, and forms interstrand cross-links in free DNA. We examined the reactivity of DOB within nucleosomes and nucleosome core particles (NCPs), the monomeric component of chromatin. Depending upon the position at which DOB is generated within a NCP, it is excised from nucleosomal DNA at a rate 275–1500-fold faster than that in free DNA. The half-life of DOB (7.0–16.8 min) in NCPs is shorter than any other abasic lesion. DOB’s lifetime in NCPs is also significantly shorter than the estimated lifetime of an abasic site within a cell, suggesting that the observed chemistry would occur intracellularly. Histones also catalyze DOB excision when the lesion is present in the DNA linker region of a nucleosome. Schiff-base formation between DOB and histone proteins is detected in nucleosomes and NCPs, resulting in pyrrolone formation at the lysine residues. The lysines modified by DOB are often post-translationally modified. Consequently, the histone modifications described herein could affect the regulation of gene expression and may provide a chemical basis for the cytotoxicity of the DNA damaging agents that produce this lesion.
Graphical abstract
INTRODUCTION
Abasic sites (AP, C4-AP, L, DOB) are produced by a variety of cytotoxic DNA damaging agents.1 Their structures, and in some instances the mechanisms for their formation, have been established by elegant studies.2–7 Elucidating the biochemical consequences of these lesions provides insight into the chemical basis for the cytotoxicity of the agents that produce them.8,9 For instance, AP, C4-AP, and L are highly mutagenic, and more recently interesting aspects of their chemical reactivity have been uncovered.10–14 For example, AP, C4-AP, and DOB yield DNA interstrand cross-links.15–21 Some of these cross-links are misrepaired by bacterial nucleotide excision repair in vitro and converted into double strand breaks.22,23 In addition, the oxidized abasic lesions, C4-AP, DOB, and L inactivate base excision repair enzymes.24–27 Histone proteins that make up chromatin also catalyze excision of the electrophilic lesions, AP, C4-AP, and L.28–31 Herein, we describe the histone-catalyzed excision of 5′-(2-phosphoryl-1,4-dioxobutane) (DOB) in nucleosome core particles (NCPs) and nucleosomes. Histone-catalyzed excision of DOB is the most rapid process yet characterized involving these proteins and studies in NCPs and nucleosomes reveal the dynamic nature of the histone tail interactions with DNA.

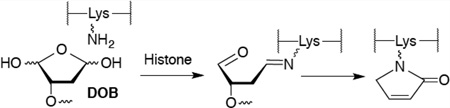
Histone proteins are at the heart of chromatin in which nuclear DNA is condensed. Chromatin is composed of NCPs that are linked via 10–90 base pairs (bp) of DNA.32 The NCPs consist of approximately 145 bp of DNA wrapped around an octameric core of highly positively charged histone proteins. The register of each base pair with respect to the histone octameric core can be referred by its superhelical location (SHL) relative to the dyad position (SHL 0), where the twofold axis of the octamer is (Figure 1A). Each turn of the duplex corresponds to one unit change in the SHL. The histone proteins contain lysine-rich, unstructured tails that protrude from the core and interact with the DNA. Post-translational modifications (PTMs) of the histone tails play an important role in regulating genetic expression.33–35 Recently, the histone tails have been shown to catalyze the excision of a variety of abasic sites (AP, C4-AP, L).28–31,36 Strand scission is accelerated as much as almost 500-fold compared to that in free DNA. For instance, at SHL 4.7, the half-life of C4-AP is 14 min.31 The lysine residues on the tail from histone H4 play a significant role in catalyzing the strand cleavage in the vicinity of SHL 1.5 by not only forming a Schiff-base with C4-AP but also facilitating β-elimination of C4-AP.31 In addition, histonecatalyzed cleavage at C4-AP results in modification of the lysine residue with an electrophilic lactam (1, Scheme 1) that contains the same charge and oxidation state as an acetylated lysine.
Figure 1.

General structure features of a nucleosome core particle. (A) Structural representation of one-half of the NCP (PDB: 1kx5). Modified nucleotides are shown as red spheres. (B) Amino acid sequence of histone H4 tail. (C) Amino acid sequence of histone H3 tail.
Scheme 1.
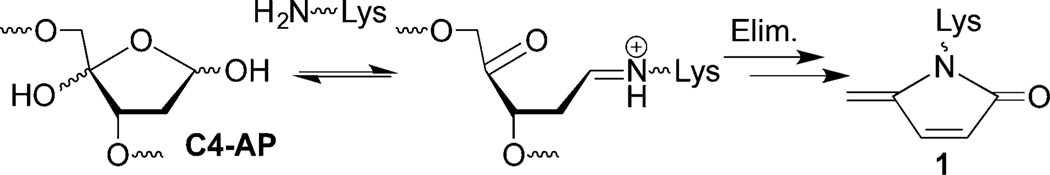
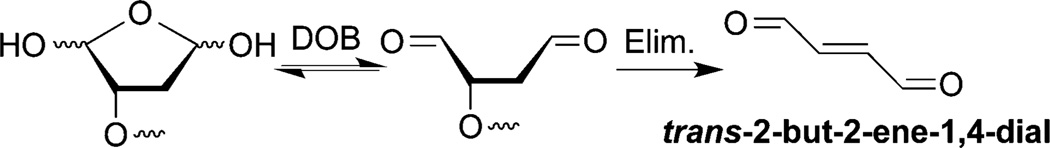
DOB is a minor product of DNA oxidation resulting from hydrogen atom abstraction from C5′ of the DNA sugar backbone.37 It is formed at the 5′-terminus concomitantly with a strand break, making it unique among abasic lesions. When formed by neocarzinostatin, the 3′-terminus of the proximal 5′-fragment contains a labile formyl phosphate, which hydrolyzes to a 3′-phosphate.7,38 Because it contains a β-phosphate leaving group, DOB undergoes β-elimination to form highly reactive cis- and trans-but-2-ene-1,4-dial (Scheme 2).39,40 In free DNA DOB forms interstrand cross-links (ICLs) selectively with dA opposite its 3′-adjacent nucleotide.21 DOB also irreversibly inhibits DNA polymerases β and λ, two enzymes that are involved in base excision repair. The lesion inactivates the lyase activities of the enzymes by reacting with lysines involved in Schiff-base formation.27,41 In this study, we investigated the reactivity of DOB at specific sites within NCPs and for the first time within nucleosomes. Including linker DNA while investigating the reactivity of DNA lesions in a nucleosome provides additional complexity that is present within the cell.
Scheme 2.
RESULTS AND DISCUSSION
Design and Preparation of NCPs and Nucleosomes Containing DOB
DOB reactivity was examined in NCPs and nucleosomes containing the 601 strong positioning sequence by independently generating the lesion at specific sites from 2.42,43 The 601 sequence binds the histone octameric core strongly, and the structure of the corresponding NCP was characterized by X-ray crystallography.44 The precursor was incorporated at three specific sites within the core particle (Figure 1A). DOB was generated within the core particle at SHL 1.5 (DOB89), SHL 4.5 (DOB119) and the dyad axis (SHL 0, DOB73). The 5′-adjacent DNA fragments were prepared containing 3′-hydroxyl groups instead of the phosphate termini likely resulting from treatment of DNA with damaging agents such as neocarzinostatin. The DNA at SHL 1.5 is bent, is a preferred binding site for DNA-damaging reagents and is close to the lysine-rich N-terminal tail of histone H4.45–47 DNA kinking in the region around SHL 4.5 severely disrupts base stacking.44 SHL 4.5 is also in close proximity to the N-terminal tails from histone H2A and H2B. The DNA is held more tightly at the dyad region (SHL 0), but is not as closely positioned to any N-terminal histone tails as at SHL 1.5 and 4.5.44,48
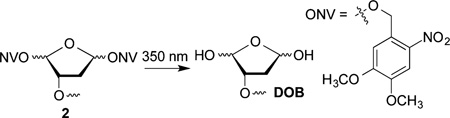
Because the strand containing DOB is already cleaved, analyzing DOB reactivity in the ≥145-bp duplexes within NCPs is technically more difficult than analysis of comparable reactivity of AP, L, or C4-AP. The strands containing latent DOB necessitate internal radiolabeling. In addition, restriction enzymes were applied following the reaction to facilitate separating DNA strands containing DOB from those without it (Scheme 3). Hence, 32P-radiolabeling and the restriction site were <17 bp from the DOB lesion.
Scheme 3.

The requisite DNA strands were PAGE-purified from the reaction in which chemically synthesized oligonucleotides were ligated.49 Hybridization of the strand containing DOB and the flanking sequence with the complementary strand yielded the requisite ternary complexes, which were reconstituted with purified octamer prepared from histone proteins expressed in E. coli to afford nucleosomes with 2 at specific sites.50 Hydroxyl radical footprinting verified that the modified nucleotide had no effect on the register of the DNA with respect to the histone octameric core (Figures S11–S15).49
To prepare nucleosomes, the 601 sequence was extended on both ends to accommodate the 46-bp linker DNA on each end whose sequence was randomly designed (sequence in Figure S1)49 DNA strands for nucleosomes from which DOB were generated at positions 73 (DOB73) and 89 (DOB89) within the core region were prepared in an analogous manner.49 The unselective cleavage pattern by hydroxyl radical observed at the first ∼40 nucleotides (nt) in the nucleosomes confirmed that the two linker regions were unshielded and that the 601 sequence fully encompassed the histone octamer (Figure S11–S15).49 Finally, DOB was incorporated at two sites within the linker region that were 13 bp (DOB176) and 30 bp (DOB159) away from the end of nucleosomal DNA (Figure 2).
Figure 2.

Generic structure of nucleosomes containing DOB in the linker DNA region.
DOB Reactivity in NCPs
DOB excision from nucleosomal DNA exhibited first-order kinetics (Figure S16). The half-life of DOB in NCPs depended upon its position, but was greatly reduced in general compared to free DNA (Table 1). Among the three positions investigated, DOB at SHL 1.5 (DOB89) exhibited the shortest half-life of 7–8.5 min, depending upon the flanking sequence, indicating that the latter has little effect on the lesion’s reactivity in NCPs. The translational position has little effect on the DOB half-life, which vary only ∼2-fold. We did not examine the effect of rotational position within a single helical turn on DOB reactivity. A previous study on AP reactivity in NCPs indicated that the effect of rotational position was modest (<3-fold), and considerably smaller than the effect observed on deamination rates of photodimers containing 5-methyl-2′-deoxycytidine.36,51
Table 1.
Decomposition Kinetics of DOB as a Function of Position within NCPs
| DOB position (SHL) |
local sequencea |
kdec (×10−3s−1) in NCPb |
t½ (min) in NCPb |
t½ (h) in free DNAc |
|---|---|---|---|---|
| 89 (1.5) | TXA | 1.4 ± 0.2 | 8.5 ± 1.1 | 119 ± 6 |
| 73 (0) | AXC | 0.7 ± 0.04 | 16.8 ± 0.8 | 77 ± 5 |
| 119 (4.5) | TXA | 0.7 ± 0.1 | 16.5 ± 1.9 | 133 ± 6 |
| 89* (1.5) | TXT | 1.6 ± 0.1 | 7.0 ± 0.5 | 179 ± 7 |
X = DOB.
Rate constants are averages ± SD of three experiments, each consisting of three independent reactions.
Data were for a single experiment consisting of three independent reactions.
indicates different local sequence for DOB89.
The rate constants for DOB decomposition in NCPs represent between an ∼275- and 1500-fold shorter lifetime than that in the corresponding free DNA. The rates of disappearance of DOB at SHL 4.5 (DOB119) and DOB73 (SHL 0) are only approximately 2-fold slower than DOB89. The variation of DOB reactivity between these four substrates is small compared to the overall acceleration observed in NCPs (∼275–1500-fold), suggesting that the presence of the lesion in the NCP is more important than its rotational orientation. It is unsurprising that ICL formation was not detected by denaturing PAGE, because the rate constant for ICL formation in free DNA (kICL = 2.6 × 10−5 s−1) is ∼100-fold slower than kdec in the NCP.21
The half-life of DOB is the shortest among all the abasic lesions (C4-AP, AP, and L) in 601 NCPs. Its accelerated reactivity in NCPs is more similar to that experienced by C4-AP (up to ∼550-fold)31 than for either AP (∼100-fold)28,29 or L (up to ∼43-fold).30 Interestingly, a plot of the rate constants for decomposition of various abasic sites at position 89 in NCPs (kdec) versus reaction in free DNA (kDNA) shows good linearity for AP, C4-AP and DOB, but not L (Figure 3). We suggest that this indicates that AP, C4-AP, and DOB react via a common mechanism (e.g., Schemes 1 and 5) but the lactone (L89) does not react via a Schiff-base intermediate.28,31
Figure 3.

Reactivity of DNA abasic sites at position 89 in NCPs within 601 DNA compared to in free DNA of the same sequence.
Scheme 5.

DNA–Protein Cross-Links
Sodium borohydride quenching, followed by SDS PAGE analysis indicated that DOB generates transient DNA–protein cross-links (DPCs), as do AP and C4-AP (Figure 4, Figure S26 and S27).28,29,31,36 The maximum DPC yield was ∼30% for DOB89 and DOB73, and ∼20% for DOB119 (Figure 4A). The maximum DPC yields were reached during the first half-life of the reactions (<5 min), and decreased rapidly thereafter (Figure 4A). DPC formation is proposed to result from Schiff-base formation between the lesion and lysine residues in the histone proteins (Scheme 4). The transient DPC formed by DOB resembles that from C4-AP89, which reached a maximum yield of <20% in 40 min.31 In contrast, the DPC formed by AP at the same site persisted for more than 24 h.28,29
Figure 4.

Time-dependent DPC formation from DOB at three positions within NCPs in the absence (A) or presence of (B) NaBH3CN.
Scheme 4.

The intermediacy of DPCs in DOB excision was probed further using NaBH3CN, which reduces Schiff-bases to alkylamines (3 and 4, Scheme 4). On account of rapid DOB reactivity, NCPs were photolyzed (5 min) in the presence of NaBH3CN (100 mM) and the reactions were monitored by SDS PAGE. The DPCs were formed in slightly higher yields (40–50%) compared to when the reducing agent was absent, and the yields remained relatively unchanged over time for DOB89 and DOB119 (Figure 4B). DOB73 produced DPCs in the highest yield (>60%) among the three DOB sites, but after reaching a maximum at 5 min they gradually decreased to a comparable level as at the other sites. The decomposition of reduced DPCs formed at DOB73 could be attributed to the β-elimination from 4 (Scheme 4). In addition, considering that DOB lesions are excised completely within 1 h, the relatively low DPC yield (40–60%) suggests that trapping by NaBH3CN was incomplete and/or that Schiff-base formation is not the sole mechanism for DOB excision.
Identification of Histone(s) Involved in DPC Formation
The DPCs formed from DOB in the presence of NaBH3CN were separated by SDS PAGE and the histone proteins cross-linked to DOB were subjected to mass spectrometry analysis following in-gel protease digestion. The identities of cross-linked proteins were confirmed by comparing the mass spectra with those obtained from wild-type (WT) histone proteins. For instance, the same characteristic fragments obtained from the single DPC band produced in a NCP containing DOB89 (Figure 5B) and the WT histone H4 (Figure 5A) indicated that DOB89 mainly cross-linked with this protein. This observation is consistent with the crystal structure that reveals that the N-terminal tail from histone H4 protrudes between the two DNA gyres in close proximity to position 89 (Figure 6).44 In addition, studies on the reactivity of AP and C4-AP in NCPs indicated that the H4 tail predominantly interacts with the abasic sites at the same position.28,29,31
Figure 5.

MALDI-TOF mass spectra of peptide fragments obtained from in-gel acetylation and trypsin digest of (A) WT histone H4 and (B) the DPC produced by DOB89. (The range of amino acids corresponding to each peptide is in parentheses.) (C) Peptide sequences and respective calculated m/z.
Figure 6.

Proximity of histone H4 tail to nucleotide 89 in the vicinity of SHL 1.5. Proximal lysine residues are shown as spheres (PDB: 1kx5).
In contrast, one major and two minor bands were observed on SDS PAGE gels from reactions of NCPs containing DOB119 (Figure S19), indicating that multiple proteins form cross-links with the lesion at this position. However, only the protein in the major band could be identified, and it was determined to be histone H2A, whose N-terminal tail is in the vicinity of position 119 (Figure S17).44,49 The reactions on NCPs with DOB73 yielded multiple DPC bands (Figure S19). Attempts to identify the cross-linked proteins were unsuccessful. We attribute this to the broader distribution of cross-linked proteins that yielded quantities of individual products below the detection limit of our mass spectrometer.
Role of Histone H4 Lys Residues in Catalyzing DOB89 Reactivity
To probe the role of specific residues on the histone H4 tail (Figure 1B) in reacting with DOB89, the reactions were carried out on NCPs containing various histone H4 variants. Substituting five lysine residues in the H4 tail (Figure 1B) with arginines, which retained the overall charge of the tail but reduced its basicity and nucleophilicity, decreased the rate of DOB decomposition within the nucleosomes containing DOB89 by ∼8-fold (Figure 7). When all of the nucleophilic residues in the histone H4 tail were mutated (Lys5,8,12,16,20 to Arg and His18 to Ala), the reactivity of DOB89 was further reduced, with the half-life ∼15 times longer than that with WT histone proteins. However, this leaves the majority (∼50-fold) of the accelerated DOB excision unaccounted for. This is in contrast to the reactivity of AP89 where histone H4 variants reveal that this protein’s tail is responsible for all but ∼5-fold acceleration of an overall ∼100-fold increase in reactivity within NCPs.29,36 However, the inability to quench the acceleration of DOB excision by removing nucleophilic/basic residues from the proximal histone H4 tail is similar to what was observed when these same experiments were carried out on NCPs containing C4-AP89.31 DOB and C4-AP share a common 1,4-dicarbonyl motif and both are significantly more reactive in NCPs than AP (Figure 3). It is possible that factors other than Schiff-base formation contribute to the acceleration of DOB and C4-AP decomposition (see below).
Figure 7.

Effects of mutated histone H4 on the reactivity of DOB89 in NCPs.
Fate of DOB89
Schiff-base formation decreases the pKa of the proton at C2 of DOB, facilitating the β-elimination (Scheme 5). Based upon previous reports we hypothesized that DOB was transferred in its entirety to the histone protein upon the elimination at its 3′-phosphate.25,31,41 To test this proposal, the four histone proteins were purified by HPLC following incubation of NCP containing DOB89 and characterized by mass spectrometry. Consistent with DPC formation (Figure 5), only modification of histone H4 was detected. This protein contained two additional ions, which were 66 and 84 Da greater than the unmodified protein (Figure S21).49 The differences in molecular weight were consistent with the formation of adducts 6 and 5 (Scheme 5), respectively. To achieve higher resolution, the purified H4 obtained from the NCP containing DOB89 was subjected to protease digestion. MALDI-TOF MS analysis following thermolysin digestion, which cleaves proteins at the amino termini of hydrophobic residues, revealed that the 20-amino acid N-terminal tail was the only region modified. Two fragments (amino acids 1–9 and 10–20) whose masses were increased by 66 Da compared to the unmodified ones were observed (Figure S22).49 Trypsin digestion following the acetylation of lysine residues cleaved at the carboxyl group of unmodified lysine and arginines. This treatment generated two modified peptide fragments (amino acids 20–23 and 4–17, Figure S23).49 The increased mass (24 Da) was consistent with the mass difference between 6 and an acetyl group, suggesting that Lys 20 and at least one of Lys 5, 8, 12, and 16 were involved in the reaction with DOB. Digestion of the modified H4 protein by Lys C (which hydrolyzes specifically at the carboxyl side of unmodified lysines) gave rise to a new peak whose mass was 66 Da greater than that of the unmodified peptide fragment 13–20 (Figure 8). The absence of the corresponding fragment in WT H4 digestion (Figure S24A) was due to cleavage at unmodified Lys 16, indicating that Lys 16 was modified by pyrrolone (6) in the NCP.
Figure 8.

MALDI-TOF mass spectrum of peptide 13–20 (calcd m/z = 975.1220) of modified histone H4 protein digested by Lys C. K* = 6.
Specific modified sites were further identified via LC-MS/MS analysis of histone H4 digested by various proteases. For instance, the mass of the peptide fragment 9–16 from the Lys C digest on modified histone H4 was 66 Da greater than the fragment from the unmodified one. In addition, the fragmentation pattern detected by LC-MS/MS affirmed Lys12 as the modification site (Figure 9A, Table S1). Similarly, the mass of the fragment consisting of amino acids 20–23 was 66 Da greater than the one from WT histone H4, which was also observed from MALDI-TOF MS analysis of the in-gel trypsin digest. The fragmentation pattern of the peptide indicated modification on Lys 20 (Figure 9B, Table S1). The above results clearly indicate that DOB89 was removed in its entirety from DNA and transferred to multiple lysine residues, including K12, K16, or K20 in the N-terminal tail from histone H4. The modified fragment 1–9 from thermolysin digest (Figure S22) suggests that K5 and/or K8 are modified by pyrrolone 6. However, the specific modification site(s) cannot be determined due to the lack of LC/MS/MS information. Similar analysis of NCPs containing C4-AP89 identified K8 as the modification site in addition to K12, K16, and K20, but not K5.31,52
Figure 9.

Peptide mapping of (A) fragment 9–16 obtained from Lys C digest and (B) fragment 20–23 from trypsin digest of H4 from NCP containing DOB89. K* = 6.
The unnatural covalent modification of lysine residues in histone protein tails by DNA lesions could have deleterious consequences on cell signaling as post-translational modifications of these residues are crucial for regulating gene expression.53,54 For instance, acetylation on Lys 16 of H4 (H4K16) prevents condensation of nucleosome arrays into higher-order chromatin fiber.55,56 Pyrrolone modification 6 shields the ε-amino group’s positive charge, as does acetylation. Therefore, such a modification on H4K16 may pass a false signal downstream. Furthermore, methylation on H4K20 plays an important role in signaling DNA damage and mediating cell cycle arrest by recruiting checkpoint protein Crb2.57–59 Cell cycle arrest provides sufficient time for the repair of DNA damage before replication resumes. The pyrrolone modification (6) on H4K20 produced by DOB may block DNA damage signaling and prevent the DNA repair enzymes from localizing at the site of damage by disrupting histone H4 binding to Crb2. Failure to induce cell cycle arrest can result in replication of damaged DNA, which is highly mutagenic. Acetylated H4K12 correlates with euchromatic structure,60 and sumoylation of the same residue also disrupts chromatin compaction and oligomerization.61 Modification 6 on H4K12 may disrupt the acetylation or sumoylation of the residue, resulting in misregulation of the dynamics of chromatin structure. These are just some of the growing number of examples of lysine modifications that affect downstream biochemical events.33,34 Given that pyrrolone 6 is generated from a DNA lesion produced by chemotherapeutic agents, histone modification by DOB could contribute to the chemical basis for the cytotoxicity of such molecules.
Effect of Linker DNA on DOB Reactivity within the Core Region
Although the NCP is a useful model to investigate the behavior of DNA lesions in chromatin, canonical corehistone tail-DNA interactions cannot be fully realized in this structure.62,63 Consequently, we examined the reactivity of DOB73 and DOB89 in nucleosomes containing linker DNA. The half-life for DOB89 excision in the NCP and nucleosome were within experimental error of one another (Table 2). These data suggest that the linker DNA did not interrupt the interactions between DOB89 and the histone H4 tail. This observation was corroborated by examining the effect of histone H4 variants on DOB89 reactivity in nucleosomes. The mutagenesis results paralleled those from the corresponding NCP. Substituting five lysine residues in the H4 tail with arginines (K5,8,12,16,20R) reduced the DOB decomposition rate by ∼7-fold (Table 2). Moreover, substituting the K5,8,12,16,20R/H18A H4 variant in the nucleosome led to a DOB half-life that was ∼17 times longer than when the WT protein was present. The comparable decreases induced by histone H4 variants with or without linker DNA (Figure 7) suggest that histone H3 does not interact with DOB89, even in the absence of nucleophilic residues on the H4 tail.
Table 2.
Effects of Linker Regions on DOB Reactivity
| position of DOB (SHL) |
DNA sequence |
H4 variant |
t½ (min) in NCPa |
|---|---|---|---|
| 89 (1.5) | no linker | WT | 8.5 ± 1.1 |
| 89 (1.5) | with linker | WT | 6.5 ± 1.2 |
| 89 (1.5) | with linker | Lys5,8,12,16,20Arg | 44.5 ± 6.7 |
| 89 (1.5) | with linker | Lys5,8,12,16,20Arg/His18Ala | 109.8 ± 8.3 |
| 73 (dyad) | no linker | WT | 16.8 ± 0.8 |
| 73 (dyad) | with linker | WT | 10.7 ± 0.4 |
Rate constants are averages ± SD of at least two experiments, each consisting of three independent reactions.
The effect of the DNA linker region on DOB reactivity was also studied at the dyad position (Table 2). Even though no N-terminal tail from a histone protein is in close proximity to DOB73, cross-linking experiments using an electrophilic probe revealed interactions between the dyad position and histones H2A, H3, and H4.52 However, such interactions may depend on the presence of the linker region. The C-terminal region of H2A interacts with DNA near the dyad position of a NCP but switches to contact the site near the edge of the core particle in nucleosomes or oligonucleosomes in which linker DNA is present.64 Any shift of the histone tail from the dyad to the linker DNA should result in decreased DOB reactivity. However, in the presence of the linker DNA, DOB73 decomposed at a slightly higher rate than in the corresponding NCP (Table 2), making it ∼500-fold faster as compared to that in the free DNA (t½ = 90.4 ± 4.4 h). The fact that linker DNA did not decrease the DOB reactivity at the dyad suggested that the linker regions did not interrupt the interactions between histones and dyad position and/or there were no interactions between linker DNA and the histone proteins.
Reactivity of DOB within the Linker DNA
To clarify whether linker DNA interacts with any of the histone N-terminal tails, and to determine how such interaction affects the DOB reactivity, we incorporated the DOB lesion at DOB159 and DOB176 (Figure 2). DOB within the linker region decomposed more slowly than it did in the intranucleosomal DNA, although the reaction was still faster relative to that in the free DNA. For instance, DOB159, which is 13 bp away from the core domain, decomposed much more slowly (t½ = 44.4 ± 4.8 min, Figure 10) than when the lesion was within the core particle (t½ = 8.5 min for DOB89, Table 1). However, DOB159 is still excised 135 times faster than in the corresponding free DNA. (t½ = 99.6 ± 4.1 h).
Figure 10.

Effects of tailless histone proteins on the reactivity of DOB159 and DOB176.
DOB176 is 30 bp away from the core, and exhibited (t½ = 190.8 ± 12.6 min, Figure 10) 4-fold lower reactivity than DOB159. However, this was still 46-fold faster than in the corresponding free DNA (t½ = 147.1 ± 7.5 h). Excision rates for DOB in the nucleosome’s linker DNA are much closer to those observed for the lesion in the NCP core (e.g., DOB89) than they are to in free DNA, suggesting that the histone proteins contribute to reaction in this region as well. The higher rate of excision at DOB159 than DOB176 is also consistent with histone protein participation because cross-linking experiments indicate that the histone tails should interact more strongly with the region containing the former.62
To determine which histone tail(s) contributes to the increased DOB reactivity within the linker region, we monitored the DOB reactivity in nucleosomes composed of tailless histone proteins. The half-life of DOB176 was unaffected by removing either the H3 or H3 and H4 tails (Figure 10), suggesting no interactions between histone tails and the oxidized abasic site at this position. In contrast, the half-life of DOB159 was extended 2-fold (t½ = 95.4 ± 0.2 min) in the absence of the H3 tails. Deleting the histone H4 tail had no additional effect on DOB159 reactivity (Figure 10). These observations suggest that DOB159 within the linker region interacts exclusively with the tail from histone H3. The histone H4 tail was not involved in DOB159 excision, even in the absence of the H3 tail. However, the histone H3 tail accounts for only ∼2-fold of the 135-fold increase in DOB159 reactivity in free DNA. Much like when DOB (and C4-AP)31 are intranucleosomal, the factors contributing to the remaining ∼65-fold acceleration are uncertain (see below).
Direct evidence for interaction between DOB159 and histone H3 was obtained by mass spectrometry. Histone H3 was separated from the other histone proteins by SDS PAGE after incubating the nucleosome containing DOB at this position. The protein was subjected to in-gel digestion by Lys C or trypsin, followed by MALDI-TOF MS or LC-MS/MS analysis respectively (Figure 11). Digestion of the modified H3 protein by Lys C, which hydrolyzes at the carboxyl side of unmodified lysine residues, yielded a peptide fragment 1–9 (m/z 1125.88, Figure 11A) whose mass was 66 Da greater than its unmodified counterpart (data not shown). The absence of the peptide fragment 1–9 in the Lys C digestion of the WT histone H3 suggested that Lys4 (as opposed to Lys9) was the position modified (Figure S24B). Lys4 modification (H3K4) was confirmed by the LC-MS/MS analysis of the mixture obtained from trypsin digestion, in which the fragment containing amino acids 3–8 was 66 Da higher than the corresponding unmodified one. Moreover, the modified Lys4 was identified by the MS/ MS fragmentation pattern of this peptide (Figure 11B, Table S1). Although the N-terminal histone tails are not visible in the NCP crystal structures, compelling evidence indicates that histone tails adopt stable conformations upon binding DNA and such structures are altered and rigidified by PTMs. Taking the predicted α-helical structure (amino acids 16–26, Figure 1C) into account, the total length of the 37-amino acid N-terminal histone H3 tail is expected to be ∼113 Å, assuming that the tail is fully extended beyond the 10-amino acid α-helical domain (1.50 Å/amino acid for α-helix structure, versus ∼3.63 Å/amino acid for fully extended chain). 66,68 DOB176 is expected to be ∼105 Å away from the core domain (1 bp ≈ 3.4 Å). Therefore, only the three most distal H3 tail amino acids (Ala, Arg, and Thr) are expected to interact with DOB176. In contrast, amino acids 1–21 (∼47.4 Å away from the core) in histone H3 tail are expected to reach DOB159, which is ∼47.6 Å from the edge of core particle. Hence, besides Lys 4, Lys 9, 14, and 18 could potentially interact with DOB159. However, only modification of Lys4 was detected.
Figure 11.

Modification of Lys4 by DOB159. (A) MALDI-TOF mass spectrum of modified histone H3 digested by Lys C (calcd m/z = 1125.297). (B) LC-MS/MS analysis on fragment 3–8 obtained from trypsin digest of modified histone H3. K* = 6.
Post-translational modification of H3K4 plays multiple roles in regulating genetic expression. For instance, trimethylated H3K4 modulates the proper transcription of the gene (MET16) that encodes for a 3′-phosphoadenylsulfate reductase in yeast by recruiting Isw1p, an ATP-dependent chromatin remodeler. Methylated H3K4 also associates with acetyl-transferase complexes, which indicates that it has a role in transcriptional activation.70 In contrast, the trimethylated H3K4 also serves as a binding platform for the recruitment of histone deacetylase complexes to induce the “closed” form of chromatin in response to DNA damage.71 The seemingly conflicting functions imply that methylated H3K4 is involved in multiple aspects of the histone code for regulating cell signaling. Given the complex multifunctional role that methylated H3K4 has, the pyrrolone modification (6) on this residue, which does not resemble any methylated form of lysine in terms of shape or charge may disrupt the recruitment of proteins and adversely affect cell signaling.
Other Factors Contributing to Accelerated DOB Reactivity in Nucleosomes and NCPs
Kinetic studies on AP89 utilizing WT and mutant variants of histone H4 revealed that the Lys (5) and His residues in the tail region accounted for all but ∼5-fold of the ∼100-fold acceleration of the lesion’s reactivity in NCPs. In contrast, ∼50-fold of the accelerated reactivity of DOB89 was unaccounted for in NCPs composed of K5,8,12,16,20R/H18A H4 (Table 1, Figure 7). Similarly, DOB176 undergoes β-elimination 46-fold faster in the nucleosome than in the corresponding free DNA However, mutagenesis and mass spectrometry experiments provided no evidence for direct histone interactions with this position in the linker DNA (Figure 10). These observations suggest that factors other than intranucleosomal protein interactions contribute to the accelerated DOB reactivity
We investigated whether random interactions between unbound histone octamer present in solution could account for part of the increased DOB reactivity. Consequently, excess carrier DNA (salmon sperm DNA) was added during nucleosome reconstitution as an attempt to remove any free histone octamers from the nucleosome solution. When the carrier DNA was in 1-fold excess of the histone octamers, the half-life of DOB176 (t½ = 4.1 ± 0.1 h) increased compared to that without the additional salmon sperm DNA (t½ = 3.2 ± 0.2 h). However, the reactivity was not affected further when 2-fold excess of carrier DNA to the histone octamer was present (t½ = 4.5 ± 0.2 h). In a complementary approach, the nucleosome solution was diluted following reconstitution to weaken any internucleosomal interactions. Maintenance of the integrity of the nucleosomes upon dilution was verified by native PAGE (Figure S28). The reactivity of DOB176 in the nucleosome was reduced by ∼3-fold (t½ = 8.9 ± 0.3 h) upon diluting the solution 10-fold. These results suggest that intermolecular interactions between free histone octamers and nucleosomal DNA account for a small portion of the accelerated DOB excision rate within the nucleosomes. However, most of the ∼50-fold acceleration of DOB reactivity still remains unaccounted for.
For comparison, AP was incorporated at position 176. The requisite duplex DNA containing precursor was prepared as described previously. In contrast to DOB, nucleosomal DNA containing AP176 underwent cleavage (t½ = 352 h, Figure S29) only ∼3-fold faster than AP sites do in free 145-bp DNA (t½ ≈1000 h).29 his comparison suggests that accelerated DOB reactivity in nucleosomes that is not accounted for by specific histone interactions is due to chemical properties that are distinct from AP.
CONCLUSIONS
5′-(2-Phosphoryl-1,4-dioxobutane) (DOB) is a lesion produced by a variety of exogenous DNA damaging reagents including ionizing radiation and the enediyne family of anticancer antibiotics. We observed that DOB is far more reactive in nucleosomes and nucleosome core particles (NCPs) than it is in free DNA. DOB reacts as much as 1,500-fold more rapidly in nucleosomes, depending upon the site where it is generated. The DOB half-life is on the order of minutes in all instances, making it the most reactive abasic lesion yet reported on in nucleosomes or NCPs. Given the repair rate of AP in cells and the fact that DOB inactivates the repair enzymes Pol β and λ, the reaction of DOB in nucleosomes and NCPs likely competes with repair of the lesion. 25,27,41,73 The electrophilicity of DOB leads to rapid reaction with proximal histone tails, resulting in transient DNA-protein cross-links (DPCs). The accelerated DOB reactivity within nucleosomes and NCPs partially results directly from these reactions. Moreover, such interactions lead to an unnatural modification (6) of the lysine residues within histone tails. The short half-life for DOB in nucleosomes may contribute to the lack of detection of the lesion in cells. These experiments suggest that searching for histone modification 6 may be a more fruitful exercise for indirectly detecting intracellular DOB production.
If formed in cellular DNA, modifications such as 6 could disrupt the homeostasis of post-translational modifications (PTMs). Disrupting PTMs in chromatin has drastic effects, resulting in various cancer types and muscular dystrophy.74–76 The pyrrolone modification on lysine residues (6) yields the same lysine side chain charge as acetylation. However, its structure is dissimilar with any natural modification on lysines. Thus, this unnatural modification may interfere with the binding of proteins on lysine residues, which could disrupt cell signaling. These effects underscore the potential importance of the discovery that DOB generates an unnatural modification on specific lysine residues in histone tails, as it may provide connection between the cytotoxicity of anticancer drugs and the DNA damage they produce.
This report also provides the first description of the reactivity of an abasic lesion in the linker DNA of a nucleosome. The greater accessibility of linker DNA in chromatin can result in preferential damage in this region by DNA damaging agents.45,77 DOB is excised more slowly within the linker region of nucleosomes than it does in the core domain of nucleosomes, but is still 46–135-fold more reactive than in free DNA. Kinetic measurements using histone variants and mass spectrometry data corroborate the interactions between the histone H3 tail and the DOB lesion in the linker region located 13 nt away from the core particle. Such interactions are partially responsible for its increased reactivity and also yield the pyrrolone modification (6) in the histone H3 tail. These modifications also have potentially significant biological ramifications and it is important to determine whether they are produced in cells.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for generous financial support from the National Institute of General Medical Sciences (GM-063028). L.W. is grateful for the Ada Sinz Hill Fellowship from Johns Hopkins University.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b05478.
General experimental methods, ESI-MS and MALDI-TOF MS of modified nucleic acids and peptides, and representative autoradiograms (PDF)
AUTHOR INFORMATION
The authors declare no competing financial interest.
REFERENCES
- 1.Dizdaroglu M. Mutat. Res., Rev. Mutat. Res. 2015;763:212–245. doi: 10.1016/j.mrrev.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Rabow LE, Stubbe J, Kozarich JW. J. Am. Chem. Soc. 1990;112:3196–3203. [Google Scholar]
- 3.Rabow LE, McGall GH, Stubbe J, Kozarich JW. J. Am. Chem. Soc. 1990;112:3203–3208. [Google Scholar]
- 4.Aso M, Kondo M, Suemune H, Hecht SM. J. Am. Chem. Soc. 1999;121:9023–9033. [Google Scholar]
- 5.Sugiyama H, Xu C, Murugesan N, Hecht SM. J. Am. Chem. Soc. 1985;107:4104–4105. [Google Scholar]
- 6.Kappen LS, Goldberg IH. Biochemistry. 1989;28:1027–1032. doi: 10.1021/bi00429a016. [DOI] [PubMed] [Google Scholar]
- 7.Chin DH, Kappen LS, Goldberg IH. Proc. Natl. Acad. Sci. U. S. A. 1987;84:7070–7074. doi: 10.1073/pnas.84.20.7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenberg MM. Acc. Chem. Res. 2014;47:646–655. doi: 10.1021/ar400229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenberg MM. Curr. Opin. Chem. Biol. 2014;21:48–55. doi: 10.1016/j.cbpa.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loeb LA, Preston BD. Annu. Rev. Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 11.Kroeger KM, Goodman MF, Greenberg MM. Nucleic Acids Res. 2004;32:5480–5485. doi: 10.1093/nar/gkh873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kroeger KM, Kim J, Goodman MF, Greenberg MM. Biochemistry. 2004;43:13621–13627. doi: 10.1021/bi048337r. [DOI] [PubMed] [Google Scholar]
- 13.Kroeger KM, Jiang YL, Kow YW, Goodman MF, Greenberg MM. Biochemistry. 2004;43:6723–6733. doi: 10.1021/bi049813g. [DOI] [PubMed] [Google Scholar]
- 14.Huang H, Greenberg MM. J. Am. Chem. Soc. 2008;130:6080–6081. doi: 10.1021/ja801715c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Catalano MJ, Liu S, Andersen N, Yang Z, Johnson KM, Price NE, Wang Y, Gates KS. J. Am. Chem. Soc. 2015;137:3933–3945. doi: 10.1021/jacs.5b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Price NE, Johnson KM, Wang J, Fekry MI, Wang Y, Gates KS. J. Am. Chem. Soc. 2014;136:3483–3490. doi: 10.1021/ja410969x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson KM, Price NE, Wang J, Fekry MI, Dutta S, Seiner DR, Wang Y, Gates KS. J. Am. Chem. Soc. 2013;135:1015–1025. doi: 10.1021/ja308119q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Regulus P, Duroux B, Bayle P-A, Favier A, Cadet J, Ravanat J-L. Proc. Natl. Acad. Sci. U. S. A. 2007;104:14032–14037. doi: 10.1073/pnas.0706044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sczepanski JT, Jacobs AC, Majumdar A, Greenberg MM. J. Am. Chem. Soc. 2009;131:11132–11139. doi: 10.1021/ja903404v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sczepanski JT, Jacobs AC, Greenberg MM. J. Am. Chem. Soc. 2008;130:9646–9647. doi: 10.1021/ja8030642. [DOI] [PubMed] [Google Scholar]
- 21.Guan L, Greenberg MM. J. Am. Chem. Soc. 2009;131:15225–15231. doi: 10.1021/ja9061695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sczepanski JT, Jacobs AC, Van Houten B, Greenberg MM. Biochemistry. 2009;48:7565–7567. doi: 10.1021/bi901006b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh S, Greenberg MM. Biochemistry. 2014;53:5958–5965. doi: 10.1021/bi500914d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashimoto M, Greenberg MM, Kow YW, Hwang J-T, Cunningham RP. J. Am. Chem. Soc. 2001;123:3161–3162. doi: 10.1021/ja003354z. [DOI] [PubMed] [Google Scholar]
- 25.Guan L, Greenberg MM. J. Am. Chem. Soc. 2010;132:5004–5005. doi: 10.1021/ja101372c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobs AC, Kreller CR, Greenberg MM. Biochemistry. 2011;50:136–143. doi: 10.1021/bi1017667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stevens AJ, Guan L, Bebenek K, Kunkel TA, Greenberg MM. Biochemistry. 2013;52:975–983. doi: 10.1021/bi301592x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sczepanski JT, Wong RS, McKnight JN, Bowman GD, Greenberg MM. Proc. Natl. Acad. Sci. U. S. A. 2010;107:22475–22480. doi: 10.1073/pnas.1012860108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou C, Sczepanski JT, Greenberg MM. J. Am. Chem. Soc. 2012;134:16734–16741. doi: 10.1021/ja306858m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou C, Greenberg MM. J. Am. Chem. Soc. 2012;134:8090–8093. doi: 10.1021/ja302993h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou C, Sczepanski JT, Greenberg MM. J. Am. Chem. Soc. 2013;135:5274–5277. doi: 10.1021/ja400915w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andrews AJ, Luger K. Annu. Rev. Biophys. 2011;40:99–117. doi: 10.1146/annurev-biophys-042910-155329. [DOI] [PubMed] [Google Scholar]
- 33.Huang H, Lin S, Garcia BA, Zhao Y. Chem. Rev. 2015;115:2376–2418. doi: 10.1021/cr500491u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowman GD, Poirier MG. Chem. Rev. 2015;115:2274–2295. doi: 10.1021/cr500350x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller MM, Muir TW. Chem. Rev. 2015;115:2296–2349. doi: 10.1021/cr5003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sczepanski JT, Zhou C, Greenberg MM. Biochemistry. 2013;52:2157–2164. doi: 10.1021/bi3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pitié M, Pratviel G. Chem. Rev. 2010;110:1018–1059. doi: 10.1021/cr900247m. [DOI] [PubMed] [Google Scholar]
- 38.Goldberg IH. Acc. Chem. Res. 1991;24:191–198. [Google Scholar]
- 39.Gingipalli L, Dedon PC. J. Am. Chem. Soc. 2001;123:2664–2665. doi: 10.1021/ja0056421. [DOI] [PubMed] [Google Scholar]
- 40.Guan L, Greenberg MM. Aust. J. Chem. 2011;64:438–442. doi: 10.1071/CH10420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guan L, Bebenek K, Kunkel TA, Greenberg MM. Biochemistry. 2010;49:9904–9910. doi: 10.1021/bi101533a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lowary PT, Widom J. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 43.Kodama T, Greenberg MM. J. Org. Chem. 2005;70:9916–9924. doi: 10.1021/jo051666k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vasudevan D, Chua EYD, Davey CA. J. Mol. Biol. 2010;403:1–10. doi: 10.1016/j.jmb.2010.08.039. [DOI] [PubMed] [Google Scholar]
- 45.Trzupek JD, Gottesfeld JM, Boger DL. Nat. Chem. Biol. 2006;2:79–82. doi: 10.1038/nchembio761. [DOI] [PubMed] [Google Scholar]
- 46.Davey G, Wu B, Dong Y, Surana U, Davey CA. Nucleic Acids Res. 2010;38:2081–2088. doi: 10.1093/nar/gkp1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuduvalli PN, Townsend CA, Tullius TD. Biochemistry. 1995;34:3899–3906. doi: 10.1021/bi00012a005. [DOI] [PubMed] [Google Scholar]
- 48.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 49.See Supporting Information [Google Scholar]
- 50.Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. Methods Enzymol. 2003;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- 51.Song Q, Cannistraro VJ, Taylor J-S. Nucleic Acids Res. 2014;42:13122–13133. doi: 10.1093/nar/gku1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weng L, Zhou C, Greenberg MM. ACS Chem. Biol. 2015;10:622–630. doi: 10.1021/cb500737y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bannister AJ, Kouzarides T. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kouzarides T. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 55.Shogren-Knaak M, Peterson CL. Cell Cycle. 2006;5:1361–1365. doi: 10.4161/cc.5.13.2891. [DOI] [PubMed] [Google Scholar]
- 56.Shogren-Knaak M, Ishii H, Sun J-M, Pazin MJ, Davie JR, Peterson CL. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 57.Botuyan MV, Lee J, Ward IM, Kim J-E, Thompson JR, Chen J, Mer G. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Du L-L, Nakamura TM, Russell P. Genes Dev. 2006;20:1583–1596. doi: 10.1101/gad.1422606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 60.Zhou BO, Wang S-S, Zhang Y, Fu X-H, Dang W, Lenzmeier BA, Zhou J-Q. PLoS Genet. 2011;7:e1001272. doi: 10.1371/journal.pgen.1001272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dhall A, Wei S, Fierz B, Woodcock CL, Lee T-H, Chatterjee C. J. Biol. Chem. 2014;289:33827–33837. doi: 10.1074/jbc.M114.591644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Angelov D, Vitolo JM, Mutskov V, Dimitrov S, Hayes JJ. Proc. Natl. Acad. Sci. U. S. A. 2001;98:6599–6604. doi: 10.1073/pnas.121171498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stefanovsky VY, Dimitrov SI, Russanova VR, Angelov D, Pashev IG. Nucleic Acids Res. 1989;17:10069–10081. doi: 10.1093/nar/17.23.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee K-M, Hayes JJ. Biochemistry. 1998;37:8622–8628. doi: 10.1021/bi980499y. [DOI] [PubMed] [Google Scholar]
- 65.Hansen JC, Lu X, Ross ED, Woody RW. J. Biol. Chem. 2006;281:1853–1856. doi: 10.1074/jbc.R500022200. [DOI] [PubMed] [Google Scholar]
- 66.Wang X, Moore SC, Laszckzak M, Ausió J. J. Biol. Chem. 2000;275:35013–35020. doi: 10.1074/jbc.M004998200. [DOI] [PubMed] [Google Scholar]
- 67.Winogradoff D, Echeverria I, Potoyan DA, Papoian GAJ. Am. Chem. Soc. 2015;137:6245–6253. doi: 10.1021/jacs.5b00235. [DOI] [PubMed] [Google Scholar]
- 68.Potoyan DA, Papoian GA. J. Am. Chem. Soc. 2011;133:7405–7415. doi: 10.1021/ja1111964. [DOI] [PubMed] [Google Scholar]
- 69.Santos-Rosa H, Schneider R, Bernstein BE, Karabetsou N, Morillon A, Weise C, Schreiber SL, Mellor J, Kouzarides T. Mol. Cell. 2003;12:1325–1332. doi: 10.1016/s1097-2765(03)00438-6. [DOI] [PubMed] [Google Scholar]
- 70.Martin C, Zhang Y. Nat. Rev. Mol. Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 71.Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Peña P, Lan F, Kaadige MR, Lacoste N, Cayrou C, Davrazou F, Saha A, Cairns BR, Ayer DE, Kutateladze TG, Shi Y, Côté J, Chua KF, Gozani O. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ruthenburg AJ, Allis CD, Wysocka J. Mol. Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 73.Atamna H, Cheung I, Ames BN. Proc. Natl. Acad. Sci. U. S. A. 2000;97:686–691. doi: 10.1073/pnas.97.2.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zeng W, de Greef JC, Chen Y-Y, Chien R, Kong X, Gregson HC, Winokur ST, Pyle A, Robertson KD, Schmiesing JA, Kimonis VE, Balog J, Frants RR, Ball AR, Jr, Lock LF, Donovan PJ, van der Maarel SM, Yokomori K. PLoS Genet. 2009;5:e1000559. doi: 10.1371/journal.pgen.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krivtsov AV, Armstrong SA. Nat. Rev. Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 77.Kuo MT, Hsu TC. Nature. 1978;271:83–84. doi: 10.1038/271083a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.