

Abstract
Motivation: The majority of variation identified by genome wide association studies falls in non-coding genomic regions and is hypothesized to impact regulatory elements that modulate gene expression. Here we present a statistically rigorous software tool GREGOR (Genomic Regulatory Elements and Gwas Overlap algoRithm) for evaluating enrichment of any set of genetic variants with any set of regulatory features. Using variants from five phenotypes, we describe a data-driven approach to determine the tissue and cell types most relevant to a trait of interest and to identify the subset of regulatory features likely impacted by these variants. Last, we experimentally evaluate six predicted functional variants at six lipid-associated loci and demonstrate significant evidence for allele-specific impact on expression levels. GREGOR systematically evaluates enrichment of genetic variation with the vast collection of regulatory data available to explore novel biological mechanisms of disease and guide us toward the functional variant at trait-associated loci.
Availability and implementation: GREGOR, including source code, documentation, examples, and executables, is available at http://genome.sph.umich.edu/wiki/GREGOR.
Contact: cristen@umich.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The list of common genetic variants associated with complex disease continues to grow as a result of increasingly powered genome wide association studies (GWAS) (Welter et al., 2014). A large proportion of the associated variants are non-coding and it has proven difficult to identify the functional variant at loci with many variants in tight linkage disequilibrium (LD). In addition, these loci often account for only a small percentage of the trait heritability which makes any minor alteration of transcript levels difficult to detect. Although eQTLs in relevant tissues can highlight loci where variants likely impact transcription of nearby genes, fine-mapping of the causal variant is plagued by the same LD patterns that impact disease association studies. Common variation located outside of protein-coding regions modulates regulatory elements in a cell-type specific manner (Claussnitzer et al., 2014; Ernst et al., 2011; Kichaev et al., 2014; Lo et al., 2014; Maurano et al., 2012; Parker et al., 2013; Pickrell, 2014; Thurman et al., 2012; Trynka et al., 2013). Examining disease-associated variants in relation to genomic regions of functional importance can give insight into the molecular mechanisms leading to disease phenotypes, particularly when all associated variants are considered in aggregate.
Our understanding of the location of regulatory elements in the genome has expanded with the advent of chromatin immunoprecipitation followed by high-throughput DNA sequencing (ChIP-Seq) technology and the Encyclopedia of DNA Elements (ENCODE) Project (The ENCODE Project Consortium, 2012). However, it is challenging to untangle meaningful biological understanding in a systematic manner, given the diverse set of data available from hundreds of cell types and tissues. With the notion that non-coding genetic variation plays a role in transcriptional regulation via regulatory epigenomic features, we can harness these data to gain knowledge of important biological mechanisms. For example, genetic variation that impacts local chromatin or methylation states and DNA accessibility can impact transcription in a given cell. In a majority of associated genomic regions, the SNP supported by ENCODE data is a SNP in strong LD with the top reported GWAS SNP (Schaub et al., 2012). Systematic chromatin profiling has revealed that variants linked with the GWAS index variant, defined here as the most strongly associated variant, are often positioned within enhancer elements active in relevant cell types (Ernst et al., 2011). Furthermore, the overlap of particular histone methylation marks with trait associated variants is cell type-specific, suggesting that gene regulation is influenced by trait alleles in a cell type-specific manner (Trynka et al., 2013). Previous work has used chromatin profiles and other ChIP-seq experimental data to investigate GWAS variation and predict the impact of candidate variants in particular genomic regions (Boyle et al., 2012; Claussnitzer et al., 2014; Kichaev et al., 2014; Lo et al., 2014; Maurano et al., 2012; Pickrell 2014; Thurman et al., 2012; Ward and Kellis 2012). However, these methods often do not consider an appropriate control set for evaluating enrichment and do not always carefully evaluate the most relevant tissues or cell types for enrichment of trait-specific variation. Identifying causal variants and mechanisms at GWAS loci remains a universal scientific challenge.
With this motivation, we developed a statistically rigorous approach to quantify enrichment of trait-associated variants in experimentally annotated functional elements such as open chromatin states, histone marks and protein-binding sites in relevant cell types to develop a clearer understanding of the underlying regulatory mechanisms. We apply an algorithm and systematic scientific method for prioritizing functional candidate variants at genome-wide significant trait-associated loci. Our aims are threefold: (i) to elucidate the important tissue/cell types in which genetic variation impacts transcription for a particular trait, (ii) to narrow our focus of the regulatory features underlying transcription disrupted by trait-associated variants and (iii) to use positional overlap with selected regulatory domains to identify potential functional candidates at trait-associated loci. To address these aims, we evaluate genetic variation identified by GWAS for five metabolic phenotypes: blood pressure (Ehret et al., 2011) (C. Newton-Cheh and P. Munroe, unpublished data), body mass index (Locke et al., 2015), coronary artery disease (Coronary Artery Disease (C4D) Genetics Consortium, 2011; Schunkert et al., 2011), lipids (Global Lipids Genetics Consortium et al., 2013) and type 2 diabetes (Morris et al., 2012). We present GREGOR (Genomic Regulatory Elements and Gwas Overlap algoRithm), an open source tool for evaluating enrichment as a method to query the vast array of ENCODE data for the design of functional experiments, enabling scientists with non-computational backgrounds to prioritize variants and loci for functional follow-up.
2 Methods
We hypothesize that the index variant reported by GWAS is not necessarily the causal variant, owing to LD at associated regions. To account for this, we first create a list of all potential causal variants by selecting variants in strong linkage disequilibrium (LD; r2 > 0.7) with trait-associated index SNPs in whole genome sequenced samples: the 1000 Genomes Phase 1 version 2 EUR Panel (Abecasis et al., 2010). Reference data from non-European populations from the 1000 Genomes Project are also available with GREGOR for selection of LD proxies. Although many indicators of regulatory potential exist for non-coding regions, we select DNase hypersensitive sites (DHSs) as a general marker of functional importance to address our first scientific question: which cell type shows strongest enrichment of trait-associated loci? We gather data from the ENCODE Project and when experimental replicates are available, we calculate the union of DHSs derived from the same tissue (Supplementary Table S1). We then examine overlap of these potential causal SNPs with DHSs from various different tissue categories. By the same approach, we later evaluate the position of the index SNPs and their LD proxies relative to histone methylation marks and ChIP-seq transcription factor-binding sites (TFBS), as well as previously defined functional chromatin states.
We calculate the total number of trait-associated loci at which either the index SNP or at least one of its LD proxies overlaps with a regulatory region across the genome (Supplementary Fig. S1). In order to evaluate the significance of this observed overlap at each individual regulatory feature, we estimate the probability of the observed overlap of GWAS SNPs relative to expectation using a set of matched control variants. For each GWAS index SNP, we identify a set of ∼500 control SNPs randomly selected from across the genome that match the index SNP for: (i) number of variants in LD, (ii) minor allele frequency (±1%) and (iii) distance to the nearest gene. When two or more GWAS index SNPs match each other following the three criteria above, they share a set of control SNPs. We consider that the number of index SNPs within its matched control set of SNPs that overlaps a given feature follows a binomial distribution with two parameters: (i) the number of GWAS index SNPs present in the control set (1 or greater), and (ii) the proportion of SNPs within the control set or their LD proxies that physically overlaps a feature. Considering the number of index SNPs that overlaps with a feature, we compute the sum of independent binomial random variables. Then for each regulatory feature, we calculate the fold-enrichment over expectation and an enrichment P-value that represents the probability that the overlap of control SNPs represented as a cumulative probability distribution is greater than or equal to the observed overlap that we see from GWAS index SNPs (Table 1). We evaluated the performance of our method using a range of parameters including different numbers of variants in LD in the matched control sets, and matched control set size (Supplementary Fig. S2). The magnitude of enrichment is generally consistent across ranges of these parameters, and the subsequent results use r2 = 0.7 with matched control set size of > 500. P-values generated based on randomly permuted sets of non-associated matched control SNPs are highly concordant with estimated P-values (see Supplementary Methods section, Supplementary Fig. S3).
Table 1.
Formulae for P-value calculation
| Inputs | A SNP set of LD-pruned r GWAS-index SNPs |
| Regulatory regions of interest formatted as BED files | |
| m = number of control SNPs selected for each index SNP | |
| Intermediate Statistics | SNP set i (1 ≤ i ≤ r) = index SNP i and its m control SNPs |
| Si ∼ Bernoulli (pi) | |
| Outputs | s = number of SNPs that fall in regulatory regions of interest in the input GWAS-index SNPs |
| Enrichment P-value = P | |
| Expected value of | |
A SNP is considered to be falling in regulatory regions of interest if itself or any of its LD proxies has positional overlap with the regions.
We attempted to evaluate the type I error rate of our enrichment method. We tested enrichment of 50 sets of randomly selected SNPs in DHSs of different tissues. SNP sets were matched with lipid-associated SNPs on 3 properties: number of LD proxies, minor allele frequency and distance to the nearest gene. A QQ plot reveals P-values that closely follow the null uniform distribution, whereas the P-value distribution for lipid-associated variants sharply deviates from the null (Supplementary Fig. S4). Additionally, we investigated type I error by first partitioning DHSs of each tissue into genic landmark categories (Parker et al., 2013) and then randomly shuffling within each category. After re-combining the DHS categories for each tissue, we evaluated enrichment of the lipid-associated variants and again compared the results to the original P-value distribution (Supplementary Fig. S4).
3 Results
3.1 Prioritizing tissue types for five phenotypes using DHSs
Our first objective is to use available epigenomic data to identify which tissues are the most biologically relevant to the trait-specific genetic variation identified by GWAS. We evaluated enrichment of independent GWAS loci for five related phenotypes: 99 blood pressure loci (BP; 2.2% trait variance explained), 97 body mass index loci (BMI; 2.7% trait variance explained) (Locke et al., 2015), 36 coronary artery disease loci (CAD; 10% trait variance explained) (Schunkert et al., 2011), 157 lipid loci (high- and low- density lipoprotein cholesterol, total cholesterol and triglycerides; 10–12% trait variance explained) (Global Lipids Genetics Consortium et al., 2013) and 65 type 2 diabetes loci (T2D; 10.7% trait variance explained) (Morris et al., 2012). DHSs are open regions of DNA accessible to protein binding, and are important in the transcriptional activity within a given cell. ENCODE has experimentally identified DHSs using DNase-seq in hundreds of cell types. We evaluate enrichment of GWAS loci in the union DHSs of cell types derived from the same tissue (Supplementary Table S1). By testing five sets of trait-associated SNPs in DHSs of 41 tissue types, we set a Bonferroni corrected threshold for significance at P < 2.4 × 10−4.
GWAS loci were significantly enriched in DHSs of tissues that are remarkably consistent with our biological understanding of the trait (Fig. 1, Supplementary Table S2).
Fig. 1.

Enrichment of GWAS variants in DHSs. Matrix of fold enrichment for five sets of trait-associated variants (BP, blood pressure; BMI, body mass index; CAD, coronary artery disease; T2D, type 2 diabetes) in DHSs of 41 tissue groups. Bonferroni significant tissues are colored based on −log10 enrichment P-value. White indicates not significant after Bonferroni correction. The dendrogram shows the relationship between different cell types based on overlap of randomly selected regions of DNase hypersensitivity across the genome
For example, BP-associated variants are highly enriched in DHSs in cell types derived from blood vessel (P = 1.2 × 10−9; fold enrichment 1.5) and heart (P = 5.3 × 10−8; fold enrichment 1.6); CAD-associated variants in DHSs from heart (P = 2.3 × 10−5; fold enrichment 1.7) and blood (P = 5.6 × 10−5; fold enrichment 1.4); lipid-associated variants in DHSs from liver (P = 2.0 × 10−14; fold enrichment 1.6), monocytes (P = 7.1 × 10−13; fold enrichment 1.9) and blood (P = 4.7 × 10−11; fold enrichment 1.4); and BMI-associated variants in DHSs in frontal cortex (P = 8.8 × 10−5; fold enrichment 1.7). We also find enrichment of BMI-associated variants in DHSs of human olfactory neurosphere-derived cells from mucosal biopsies (P = 4.2 × 10−5; fold enrichment 1.7), suggesting a plausible link between olfaction and food intake. However, there are other cases in which we find enrichment of trait-associated variants in unexpected tissue types. For example, although we observe significant enrichment of T2D-associated variants in pancreatic tissue as expected (P = 1.0 × 10−4; fold enrichment 1.6), we see stronger evidence for enrichment in heart tissue (P = 1.4 × 10−6; fold enrichment 1.7) and embryonic stem cells (P = 2.5 × 10−6; fold enrichment 1.5). We used this knowledge to guide subsequent enrichment analysis of other epigenomic features by focusing on the most significant cell types to reduce the multiple testing burden in subsequent assessments of additional regulatory features. This data-driven approach to reduction of a large set of potentially relevant regulatory elements in a myriad of cell lines and tissues can be used for phenotypes where little is known about the biology, and may also identify novel tissues where these GWAS loci are actively transcribed. Alternatively, investigators might bypass this step and instead use a priori biological knowledge to focus on a specific tissue or cell type. One could also integrate the two approaches to choose some empirically-selected cell types but up-weight biologically relevant cell types.
We additionally investigate whether enrichment is tissue type-specific. Given the wealth of DHS data available, often in replicates and for multiple cell types from the same tissue type, we hypothesize that each cell type has some level of missing data and artifacts. To address this, we define ‘consensus’ regions of open chromatin that are commonly shared among at least 50% of all cell types within a single tissue group, and re-evaluate enrichment of lipid-associated GWAS variants. We additionally compare results for consensus thresholds (proportion of cell types required to show a DHS at that genomic position) of 100, 75, 25% and the union of cell types derived from the same tissue. We found that when we used stricter definitions to select functional regions (e.g. 100% of cell types were required to share the DHS), we typically observed higher fold enrichment, but less significant enrichment P-values (Supplementary Fig. S5, Supplementary Table S3). Conversely, when we relaxed the criterion to allow DHSs observed in only 25% of cell types, we typically observed stronger P-values but lower fold enrichment. This is likely due to inclusion of more artefactual DHSs using the relaxed definition, but exclusion of true DHSs under the strict definition. In subsequent analyses, we used the most relaxed definition of regulatory elements by including any element observed in at least one replicate or cell type within each tissue category. We opted to be more inclusive to allow for the most complete identification of DHSs.
3.2 Prioritizing regulatory elements in selected tissues
Following prioritization of important tissue types for GWAS of a specific phenotype, we next selected specific regulatory elements that were enriched for GWAS variants in cell types derived from relevant tissues, focusing solely on the tissues selected in Step 1. We evaluated enrichment of trait-associated variants in chromatin states predicted from histone methylation marks and a learned multivariate hidden Markov model (Ernst et al., 2011) (Supplementary Fig. S6, Supplementary Table S4). Confirming previous reports (Maurano et al., 2012), we found significant enrichment of genetic variation in weak and strong enhancer states for nearly all phenotypes tested. Trait-associated variants are most highly enriched in active promoters commonly marked by H3K4me2, H3K4me3, acetylation, or H2A.Z. There is less striking enrichment in domains that contain repressed genes such as H3K9me2, H3K9me3 or H3K27me3.
We further evaluated enrichment in TFBS and histone modifications identified by ChIP-Seq. We investigated any Tier 1 or 2 ENCODE cell types available for relevant tissues identified in Step 1, taking the union of experimental replicates when available. For cell types HepG2, Monocytes CD14+ (RO01746), GM12878, K562 and CD20+ (RO01778), we find significant enrichment of lipid-associated variation for key transcriptional machinery including RNA Polymerase II (P = 6.2 × 10−24; fold enrichment 2.0) and the ubiquitous transcription factor SP1 (P = 1.5 × 10−15; fold enrichment 3.0). In addition, lipid-associated variants are highly enriched in binding sites of RCOR1 (P = 1.8 × 10−16; fold enrichment 2.1), EP300 (P = 1.2 × 10−14; fold enrichment 2.0), JUND (P = 2.2 × 10−14; fold enrichment 2.0) and H3K4me3 (P = 1.4 × 10−13; fold enrichment 2.1) (Supplementary Table S5). We tested a total of 158 regulatory features, 75 of which reach Bonferroni significance with P < 3.2 × 10−4. We are particularly interested in 15 known lipid gene regulators as well as 16 transcription factors and 4 histone markers associated with lipid change in the literature. Of the 75 Bonferroni significant regulatory features, 18 of these are among this a priori-defined lipid-related list of 35 elements.
3.3 Prioritizing candidate functional variants using selected regulatory elements in relevant tissues
As we gain knowledge about the transcriptional machinery that acts in concert with trait-associated genetic variation, we can make more informed predictions about potential functional variants at a single locus. We hypothesize that variants present within multiple regulatory domains are more likely to play a role in transcriptional regulation within a cell. Subsequently, we can use this information in combination with functional protein-coding information, transcript level annotation, and deleteriousness scoring to prioritize loci and individual variants for functional follow-up.
We proceeded to prioritize potential functional variants in the 157 known lipid-associated loci (Supplementary Fig. S7). With the assumption that a protein-coding variant is likely the functional driver of transcription at a given locus, we excluded any lipid-associated loci from follow-up consideration that contains at least one non-synonymous variant in LD (r2 > 0.7) with the GWAS index SNP. This resulted in 103 remaining loci for further evaluation. We next examined our results from Step 2 to focus on the selected transcription factors and histone marks, and prioritized loci at which multiple transcription factors bind in blood, monocytes, or liver. In a data-driven approach, we flagged variants that overlap with a subset of significantly enriched regulatory domains as plausible functional candidate SNPs (n = 23). We evaluated overlap of GWAS variants at candidate loci in lipid gene regulators as well as transcription factors and histone marks involved with lipid change in the literature. Variants at a set of five of these loci that overlap with at least eight (25%) lipid-related regulatory features were commonly found using both the data-driven and biological-driven selection of regulatory features, including the known functional variant rs12740374 at SORT1 (Musunuru et al., 2010). Many of these candidate variants are also eQTLs in liver, omental fat or subcutaneous fat or had at least one surrogate SNP in LD (r2 > 0.7) with the eQTL SNP at that locus (eQTL P < 1 × 10−3) (Schadt et al., 2008). In addition to considering these various data, we counted the number of variants at each locus and focused on loci with relatively few numbers of variants to increase the likelihood of identifying the functional variant. Thus, we narrowed down the list of lipid loci that likely have a strong impact on regulating transcription to guide us to promising candidates for functional follow-up.
After analyzing the overlap of non-coding variants with biological TFBS from ChIP-seq and using our criteria of non-coding variants, eQTLs, and number of variants at a locus, we chose five loci and picked one SNP from each region that had some evidence of being the functional variant due to overlap with the most regulatory regions for further study (FAM117B: rs11692610; ANGPTL8: rs737337; SPTLC3: rs1321940; IRF2BP2: rs526936; ADH5: rs1800759) (Fig. 2, Supplementary Fig. S8).
Fig. 2.

Physical overlap of variants at the ANGPTL8 locus with experimentally defined regulatory features. SNPs within r2 > 0.7 of the GWAS index SNP overlap with ChIP-seq regions, DNase-seq binding sites or histone marks for lipid-related ENCODE features. GWAS HDL cholesterol –log10 P-values are also shown (when available) (Teslovich et al., 2010). The purple dotted line annotates the hypothesized functional variant based on physical overlap prediction (rs737337), which is also the top SNP as reported by GWAS at this locus. The blue dotted line annotates the control SNP (rs3810308). Regulatory elements highlighted in red show overlap with the purple candidate functional variant
At each locus, we selected an additional variant with no predicted C/EBP binding site overlapping as an internal control (FAM117B: rs11694172; ANGPTL8: rs3810308; SPTLC3: rs364585; IRF2BP2: rs514230; ADH5: rs2602836). Variant rs12740374 from the SORT1 locus has previously been demonstrated to alter a C/EBP TFBS (Musunuru et al., 2010), and thus was used as a positive control here (rs629301 as the SORT1 locus internal control).
We next attempted to directly determine the allele-specific effects of the non-coding SNP polymorphism on transcription factor binding at each of the six lipid loci. We generated luciferase constructs containing ±300–400 bp around each genetic variant (generating the alternate allele with site-directed mutagenesis) and transfected them into HepG2 cells over-expressing C/EBP-β. We normalized luciferase activities to the pcDNA3.1-co-transfected groups (control construct with no C/EBP-β DNA inserted), and found robust luciferase activity increase in the rs12740374-T construct compared with rs12740374-G from the SORT1 locus (fold increase = 1.8, P = 4 × 10−4), which was consistent with the previous report. Similarly, for the other five loci examined, the single nucleotide changes in other predicted functional SNP sites caused significant luciferase activity differences in response to C/EBP-β over-expression (P < 0.05), indicating those non-coding variants may change transcription factor binding activity in GWAS loci and possibly affect downstream gene expression (Fig. 3). After correction for 12 tests (2 SNPs at six loci), we still find significant differences between the two alleles (P < 0.05) at the candidate functional SNP for five out of the six loci (SORT1: rs12740374-T; ANGPTL8: rs737337-C; FAM117B: rs11692610-T; IRF2BP2: rs526936-C; SPTLC3: rs1321940-G). In contrast, the luciferase signal changes of the internal control SNP constructs were significant at Bonferroni levels for only two of the six loci.
Fig. 3.

Luciferase assays with constructs containing non-coding SNP regions at lipid loci. Relative firefly luciferase expression from constructs with haplotypes of 600–800 bp regions was transfected into HepG2 cells. Single nucleotide alterations in each variant were introduced into constructs as indicated and all luciferase activities were normalized to their pcDNA3.1 co-transfected control groups. Nominal P-values and SD (n = 8) for each SNP are shown. The PGL4 empty vector control is on the far left, while the predicted functional variant and control variant follow next in each individual locus figure
Our results are generally supported by post hoc annotations of predicted regulatory elements defined by RegulomeDB (Boyle et al., 2012). For example, the RegulomeDB score of the GWAS HDL cholesterol-associated index SNP at the SPTLC3 locus (rs364585) is 5, indicating that there is TF binding or DNase peak epigenomic data to support its functionality. In contrast, the RegulomeDB score of our predicted functional SNP at this locus (rs1321940) is 2a, indicating that it is likely to affect binding based on evidence of TF binding, and the presence of a matched TF motif, DNase footprint, and DNase peak. Among all 18 variants within r2 > 0.7 of the index SNP at this locus, only 2 SNPs have a score of 2b or better. We observe similar trends for other loci at which we performed experimental follow-up (Supplementary Fig. S8). Counterintuitively, the known functional variant rs12740374 at the SORT1 locus has a RegulomeDB score of 2b, whereas the variant reported by GWAS in that region, rs629301, has a higher score of 1f. This result emphasizes the need to consider multiple sources of data when prioritizing functional candidates for experimental follow-up. Although annotation of individual variants is useful in predicting the potential impact of a single variant, GREGOR considers all trait-associated variants in aggregate to prioritize which functional elements and in which tissues they are most relevant for the trait being examined. An alternative is considering the entirety of ENCODE data, much of which will represent irrelevant tissue types or highly correlated data sets.
4 Summary
We have developed a systematic approach for evaluating enrichment of trait-associated variants in epigenomic features, allowing us to prioritize tissues, regulatory elements, and potential functional variants that affect transcriptional regulation (Fig. 4). Our method takes into account all potential causal variants at a locus due to LD and estimates enrichment with particular regulatory features using matched control variants. It is an unbiased approach that can be used to narrow the focus of cell types and regulatory features that does not rely on a priori knowledge of biological mechanisms. The resultant findings will guide us to a more global understanding of the underlying epigenomic architecture leading to trait-specific variation.
Fig. 4.
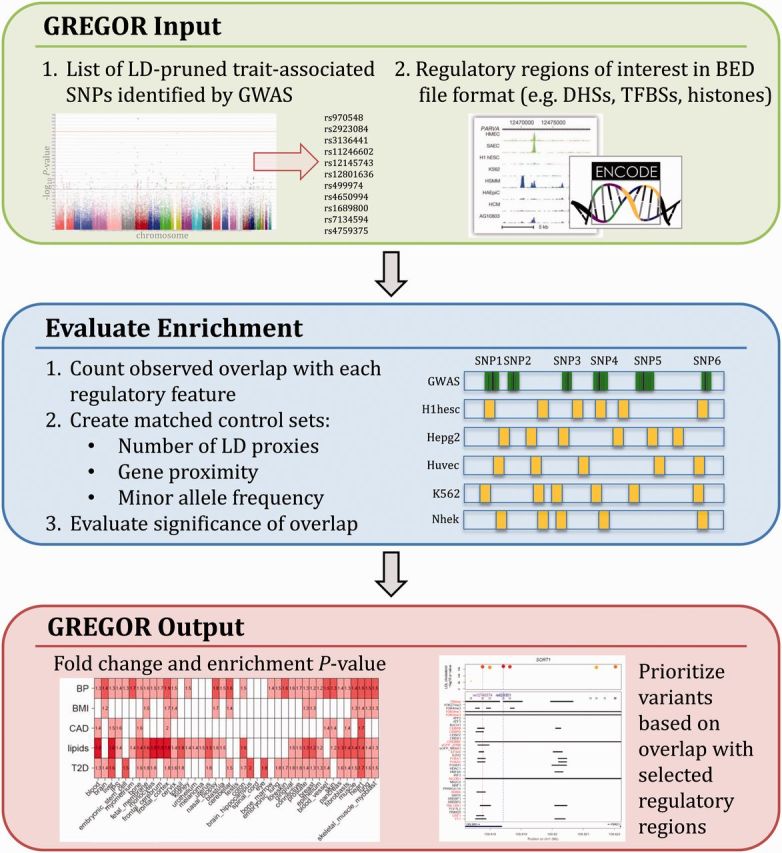
Overview of GREGOR design
We present here one reasonable approach for prioritizing the potential functional variant at a locus. We attempted to select loci with the best chance of demonstrating a functional variant for experimental follow-up. However, our approach is limited to only one potential functional variant per locus and does not claim to definitively identify the true or only functional variant at any locus. More comprehensive interrogation of variation within a locus will be required to fully understand the underlying molecular mechanisms involved.
Different cell types and tissues are more easily accessible than others for sequencing. This approach will become even more impactful as we develop an increasingly comprehensive and diverse interrogation of the epigenome to answer important biological questions about the regulatory role of non-coding variation. In all, this approach will help guide our knowledge of the important mechanisms occurring outside of protein-coding regions that underlie cell-type-specific transcriptional regulation.
Supplementary Material
Acknowledgements
The authors thank Praveen Sethupathy and Michael Stitzel for helpful discussion at the initiation of the project. We also thank Martin Buchkovich, Charles Burant, Ruth Loos, and Stephen Parker for helpful input.
Funding
This work was directly supported by HL094535 (C.J.W.) and by the National Science Foundation Open Data IGERT Grant (0903629) (E.M.S.). C.J.W. was additionally supported by HL109946 and HL127564.
Conflict of Interest: none declared.
References
- Abecasis G.R., et al. (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle A.P., et al. (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res., 22, 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claussnitzer M., et al. (2014) Leveraging cross-species transcription factor binding site patterns: from diabetes risk loci to disease mechanisms. Cell, 156, 343–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronary Artery Disease (C4D) Genetics Consortium(2011) A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat. Genet., 43, 339–344. [DOI] [PubMed] [Google Scholar]
- Ehret G.B., et al. (2011) Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature, 478, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., et al. (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature, 473, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichaev G., et al. (2014) Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet., 10, e1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo K.S., et al. (2014) Strategies to fine-map genetic associations with lipid levels by combining epigenomic annotations and liver-specific transcription profiles. Genomics, 104, 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke A.E., et al. (2015) Genetic studies of body mass index yield new insights for obesity biology. Nature, 518, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano M.T., et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science, 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris A.P., et al. (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet., 44, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K., et al. (2010) From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature, 466, 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S.C., et al. (2013) Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl Acad. Sci. U S A, 110, 17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell J.K. (2014) Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet., 94, 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt E.E., et al. (2008) Mapping the genetic architecture of gene expression in human liver. PLoS Biol., 6, e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub M.A., et al. (2012) Linking disease associations with regulatory information in the human genome. Genome Res., 22, 1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schunkert H., et al. (2011) Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet., 43, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich T.M., et al. (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature, 466, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman R.E., et al. (2012) The accessible chromatin landscape of the human genome. Nature, 489, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trynka G., et al. (2013) Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet., 45, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward L.D., Kellis M. (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res., 40, D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D., et al. (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res., 42, D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Lipids Genetics Consortium, et al. (2013) Discovery and refinement of loci associated with lipid levels. Nat. Genet., 45, 1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.