

Abstract
Viruses have developed various strategies to protect infected cells from apoptosis. HIV-1 infected macrophages are long-lived and considered reservoirs for HIV-1. One significant deciding factor between cell survival and cell death is glucose metabolism. We hypothesized that HIV-1 protects infected macrophages from apoptosis in part by modulating the host glycolytic pathway specifically by regulating hexokinase-1 (HK-1) an enzyme that converts glucose to glucose-6-phosphate. Therefore, we analyzed the regulation of HK-1 in HIV-1 infected PBMCs, and in a chronically HIV-1 infected monocyte-like cell line, U1. Our results demonstrate that HIV-1 induces a robust increase in HK-1 expression. Surprisingly, hexokinase enzymatic activity was significantly inhibited in HIV-1 infected PBMCs and in PMA differentiated U1 cells. Interestingly, we observed increased levels of mitochondria-bound HK-1 in PMA induced U1 cells and in the HIV-1 accessory protein, viral protein R (Vpr) transduced U937 cell derived macrophages. Dissociation of HK-1 from mitochondria in U1 cells using a pharmacological agent, clotrimazole (CTZ) induced mitochondrial membrane depolarization and caspase-3/7 mediated apoptosis. Dissociation of HK-1 from mitochondria in Vpr transduced U937 also activated caspase-3/7 activity. These observations indicate that HK-1 plays a non-metabolic role in HIV-1 infected macrophages by binding to mitochondria thereby maintaining mitochondrial integrity. These results suggest that targeting the interaction of HK-1 with the mitochondria to induce apoptosis in persistently infected macrophages may prove beneficial in purging the macrophage HIV reservoir.
Keywords: apoptosis, glucose metabolism; hexokinase; HIV-1; macrophage; mitochondria
Abbreviations
- HK-1
Hexokinase-1
- Vpr
viral protein R
- CTZ
Clotrimazole
- G-6-P
glucose-6-phosphate
- G6PD
glucose-6-phosphate dehydrogenase
- OMM
outer mitochondrial membrane
- VDAC
voltage-dependent anion channel
- COXIV
Cytochrome c oxidase subunit IV
- cART
combination antiretroviral therapy
- M-CSF
macrophage colony-stimulating factor
Introduction
Monocytes and macrophages play important roles in innate immune defense.1 However, in the context of HIV-1 infection, macrophages can promote viral spread from the periphery to end organs such as the brain.2 Numerous studies have demonstrated that HIV-1 infected monocytes and macrophages are relatively resistant to apoptosis.3-6 Therefore, these long-lived infected macrophages can serve as reservoirs for HIV-1 persistence in the brain and promote inflammation in the CNS of patients receiving combination antiretroviral therapy (cART).7-9 Understanding the mechanistic basis of resistance to apoptosis in HIV-1 infected macrophages is essential for therapeutic targeting of these viral reservoirs. The resistance to apoptosis of infected macrophages may stem from reciprocal interaction between HIV-1 and host glucose metabolism. Indeed, our earlier study demonstrated that Vpr modulates the glycolytic pathway in macrophages.10
The first step in glucose metabolism is the conversion of glucose to glucose-6-phosphate (G-6-P) by hexokinases (HKs).11 In mammalian cells, there are 4 isoforms of HK, HK-1, HK-2, HK-3 and HK-4.11 Among these isoforms of HKs, HK-1 and HK-2 can bind to the mitochondria.12,13 Mitochondria bound HK plays a significant role in the maintenance of the integrity of the outer mitochondrial membrane (OMM). Studies have shown that binding of HK to voltage-dependent anion channel (VDAC) inhibits the release of intermembrane proteins by apoptotic stimuli and suppresses apoptosis.14-16
The long-lived persistence of macrophages as viral reservoirs is a significant challenge in the formulation of HIV-1 eradication strategies in the current era of effective cART. Furthermore, elucidation of the mechanism involved in HIV-1-mediated hijacking of the glucose metabolism pathway might provide insights in designing therapeutic strategies for eradication of latent HIV-1.
In this study, we show that HIV-1 induces HK-1 expression in monocytes and macrophages. However, HIV-1 expression inhibited HK activity but promoted an increase in HK-1 association with the mitochondria thereby maintaining the integrity of the OMM. Our studies also show that overexpression of Vpr in U937 cell derived macrophages increases mitochondrial association of HK-1. Detachment of mitochondrial HK-1 with CTZ in HIV-1 infected macrophages decreased mitochondrial membrane potential and induced caspase-3/7 dependent apoptosis. Similarly, dissociation of HK-1 with CTZ in Vpr transduced U937 cell derived macrophages induced caspase-3/7 activity. These observations thus provide insights for the development of pharmacological therapeutic strategies for the modulation of HK-1-mitochondria interaction in infected macrophages for viral eradication by promoting apoptosis.
Results
HIV-1 induces HK-1 expression in monocytes and macrophages
We explored whether HIV-1 infection of PBMCs and reactivation of HIV-1 expression in chronically infected U1 cells modulate expression of enzymes involved in glucose metabolism.
We assessed the expression level and activity of HK that catalyzes glucose phosphorylation. Western analyses of cell lysates prepared from mock infected and HIV-1 infected PBMCs after 6 d of infection wherein the p24 levels were 71 ± 10.3 ng/ml, demonstrated a significant increase in the expression of HK-1 by about 2.4-fold but not of HK-2 (Figs. 1A, B). Since HK-1 expression was increased in HIV-1 infected cells, we therefore assessed total HK activity levels. Surprisingly, we observed a 50% decrease in total HK enzymatic activity in HIV-1 infected PBMCs in comparison to mock-infected PBMCs (Fig. 1C).
Figure 1.

HIV-1 induces HK-1 expression in monocytes/macrophages. (A) Representative Western blot of PBMCs mock infected (mock) or infected with HIV-1 SF162 (HIV-1) showing HK-1, HK-2 and α-tubulin protein expression from whole cell lysates 6 d post-infection. (B) Densitometric analysis of HK-1 expression levels normalized to tubulin levels, plotted as fold change in HK levels. Data obtained from 2 gels * indicates p value < 0.03. (C) Total HK enzyme activity in arbitrary values (represented by OD 340 nm normalized to protein concentration) in PBMCs infected with HIV-1 SF162.* indicates p value < 0.04. (D) Representative Western blot of HK-1, HK-2 and Grb2 protein expression in undifferentiated U1 cells (Con) and PMA differentiated U1 cells (PMA) cell lysates. (E) Densitometric analysis of HK-1 expression levels normalized to Grb2 levels, plotted as fold change in HK levels. * indicates p value < 0.02. (F) Total HK enzyme activity in arbitrary values (represented by OD 340 nm normalized to protein concentration) in cell lysates prepared from undifferentiated U1 cells (U1 Con) and PMA differentiated U1 cells (U1 PMA). * indicates p value < 0.01. (G) Representative Western blot of HK-1, HK-2 and Grb2 protein expression in undifferentiated U937 cells (Con) and PMA differentiated U937 cells (PMA) cell lysates. (H) Densitometric analysis of HK-1 expression levels normalized to Grb2 levels, plotted as fold change in HK levels. * indicates p value < 0.05. (I) Total HK enzyme activity in arbitrary values (represented by OD 340 nm normalized to protein concentration) in cell lysates prepared from undifferentiated U937 cells (U937 Con) and PMA differentiated U937 cells (U937 PMA) * indicates p value < 0.04.
Since bulk analyses of HIV-1 infected monocytes and macrophages limit our ability to discriminate whether changes occurred exclusively in productively infected cells, on bystander uninfected cells or in both populations, we utilized a cell line model to mimic human HIV-1 infected macrophages. The human pro-monocytic U937 cell line is one of the most commonly used HIV-1 monocyte/macrophage model system.17 U1 cells are chronically HIV-1 infected U937 cells18 and used extensively to study the effects of HIV-1 in macrophages.19-21
Reactivation of HIV-1 replication upon PMA stimulation in U1 cells (p24 levels: 8929 ± 652 ng/ml at 72 h), up-regulated expression of HK-1 by 3.6-fold, but HK-2 levels were not changed (Figs. 1D, E). However, a significant decrease (78%) in total HK enzymatic was observed after induction of viral replication with PMA in U1 cells in comparison to uninduced U1 cells (Fig. 1F). To assess whether PMA treatment inhibited total HK activity in U1 cells, U937 cells were treated with PMA in a similar fashion, and total HK enzymatic activity assay was measured. We first assessed expression levels of HK-1 and HK-2 in these lysates. PMA treatment of U937 cells led to a moderate (1.5-fold) increase in HK-1 expression with no change in HK-2 expression (Figs. 1G, H). However, we observed a 23% reduction in total HK enzymatic activity in comparison to un-induced U937 cells (Fig. 1I). Taken together, these observations show that though differentiation of U937 by PMA induces HK-1 expression, HIV-1 infection or reactivation of HIV-1 in U1 cells significantly induces HK-1 expression and down regulates HK enzymatic activity in monocytes and macrophages, respectively.
HIV-1 increases mitochondrial HK-1 levels
Studies have shown that the association of HK-1 and HK-2 with the mitochondria protects against cell death.15,22-24 Therefore, we assessed whether HIV-1 modulates translocation of HK-1 from the cytoplasm to the mitochondria in macrophages and thereby regulate survival of HIV-1 infected macrophages. Uninduced and PMA induced U1 cells were harvested after 72 h when viral replication peaks and were subjected to cytoplasmic and mitochondrial fractionation. The purity of the mitochondrial and cytoplasmic fractions was ascertained by using VDAC1 or COXIV as a mitochondrial and tubulin as a cytoplasmic marker. Levels of HK-1 in both the cytoplasm and mitochondria of U1 cells increased with viral replication in contrast to the un-induced cells in which the HK-1 was predominantly in the cytoplasm with very low amounts in the mitochondria (Figs. 2A–C). To ascertain that PMA treatment did not modulate subcellular localization of HK-1 in U1 cells, control experiments were performed with PMA induced U937 cells. The results show that PMA treatment of U937 did not lead to an increase in mitochondrial HK-1 levels though it increased total HK-1 levels (Figs. 1G, H). PMA decreased levels of mitochondrial HK-1 and increased cytoplasmic HK-1 levels in comparison to uninduced U937 cells in which there was relatively higher levels of HK-1 in the cytoplasm compared to the levels in mitochondria (Fig. 2D–F). These observations demonstrate that HIV-1 modulates subcellular localization of HK-1 favoring increased association of HK-1 with mitochondria.
Figure 2.
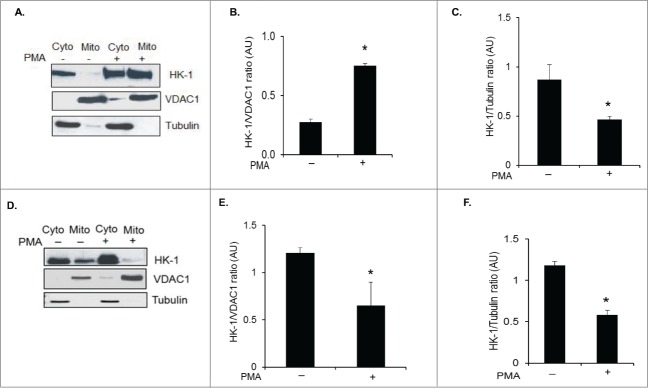
HIV-1 increases mitochondria bound HK-1 levels in U1 cells. (A) Uninduced (−) and PMA induced (+) U1 cells were harvested after 72 h of induction and fractionated into cytoplasmic and mitochondrial fractions. The cytoplasmic and mitochondrial fractions were analyzed by Western blot for the changes in expression levels of HK-1. VDAC1 and α-tubulin were used as markers for fraction purity and protein loading of mitochondrial and cytoplasmic fractions, respectively. (B) Densitometric analysis of HK-1 expression levels in the mitochondrial fraction normalized to VDAC1 levels in uninduced (−) and induced cells (+), shown as HK-1/VDAC1 ratio on the y-axis in arbitrary unit (AU). * indicates p value < 0.02. (C) Densitometric analysis of HK-1 expression levels in the cytoplasmic fraction normalized to α-tubulin levels in uninduced (-) and induced (+) U1 cells, shown as HK-1/Tubulin ratio on the y-axis in arbitrary unit (AU). * indicates p value < 0.05. (D) Uninduced (-) and PMA induced (+) U937 cells were harvested after 72 h of induction and fractionated into cytoplasmic and mitochondrial fractions. The cytoplasmic and mitochondrial fractions were analyzed by Western blot for the expression of HK-1. VDAC1 and α-tubulin were used as markers for fraction purity and protein loading of mitochondrial and cytoplasmic fractions, respectively. (E) Densitometric analysis of HK-1 expression levels in the mitochondrial fraction normalized to VDAC1 levels in uninduced (-) and induced (+) U937 cells, shown as HK-1/VDAC1 ratio on the y-axis in arbitrary unit (AU). * indicates p value <0.05. (F) Densitometric analysis of HK-1 expression levels in the mitochondrial fraction normalized to α-tubulin levels in uninduced (-) and induced (+) U937 cells, shown as HK-1/Tubulin ratio on the y-axis in arbitrary unit (AU). * indicates p value <0.03.
Cytoprotective role of mitochondrial HK-1 in HIV-1-infected cells
To elucidate the protective role of mitochondria associated HK-1 in HIV-1-infected cells, we used the anti-fungal imidazole derivative, clotrimazole (CTZ) that has been shown to release mitochondria bound HK and induce apoptosis.15,22,26,27 Since CTZ dissociates HK once it is bound to mitochondria and not inhibit HK from binding to the mitochondria in the following experiments cells were treated with CTZ post treatment with PMA and not pretreated with the agent. U1 cells were induced by PMA, and 72 h post induction cells were treated with 2 different concentrations of CTZ (10 and 25 μM) or DMSO as vehicle control for 10 h and HK-1 protein levels were assessed in cytoplasmic and mitochondrial fractions. This analysis demonstrates that the ratio of the HK-1 in the cytoplasmic fraction (Figs. 3A, B) versus the mitochondrial fraction (Figs. 3C, D) was modulated in a dose-dependent manner by CTZ. Specifically, CTZ at 25 μM significantly increased cytoplasmic levels of HK-1 (Figs. 3A, B) with a concomitant decrease in HK-1 levels in the mitochondrial fraction (Figs. 3C, D). In subsequent experiments, we used CTZ at a concentration of 25 μM.
Figure 3.
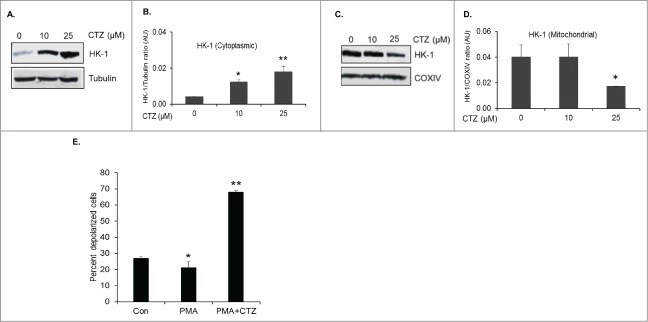
Dissociation of mitochondria bound HK-1 by Clotrimazole leads to mitochondrial depolarization. (A) Representative Western blot of HK-1 and α-tubulin levels in the cytoplasmic fraction of PMA differentiated U1 cells treated with 10 and 25 μM of CTZ. (B) Densitometric analysis of HK-1 levels in the cytoplasmic fraction normalized to α-tubulin levels, shown as HK-1/Tubulin ratio on the y-axis in arbitrary unit (AU). *, ** indicate p values <0.03 and <0.02, respectively in comparison to no treatment with CTZ. (C) Representative Western blot of HK-1 and COXIV levels in the mitochondrial fraction of PMA differentiated U1 cells treated with 10 and 25 μM of CTZ. (D) Densitometric analysis of HK-1 levels in the mitochondrial fraction normalized to COXIV levels, shown as HK-1/COXIV ratio on the y-axis in arbitrary unit (AU). * indicates p value <0.02 in comparison to no treatment with CTZ. (E) Assessment of changes in mitochondrial membrane potential in response to CTZ. Mitochondrial depolarization was assessed using membrane permeable dye, JC-1 in uninduced U1 cells (control), PMA differentiated U1 cells (PMA), and PMA differentiated U1 cells treated with 25 μM CTZ (PMA+25 μM CTZ). A 5% decrease in depolarized cells in PMA alone treated cells in comparison to control cells * indicates p value <0.05 Significant increase in percentage of depolarized cells was observed in PMA+ 25 μM CTZ treated cells in comparison to PMA alone treated cells. ** indicates p value <0.01.
To assess the effects of changes in the subcellular localization of HK-1 by CTZ (25 μM) on mitochondrial fitness, we measured changes in mitochondrial membrane potential (ΔΨ) using the membrane permeable dye, JC-1. The percentage of polarized cells (red fluorescence) in control U1, PMA treated U1 cells and PMA plus 25 μM CTZ treated U1 cells were 72.39 ± 1.6%, 73.98 ± 2.8% and 31.71 ± 1.3%, respectively. The percentage of depolarized cells (green fluorescence) in control U1, PMA treated U1 cells and PMA plus 25 μM CTZ treated U1 cells were 26.83 ± 1.2%, 21.28 ± 3.5% and 68.05 ± 1.0%, respectively (Fig. 3E). These studies demonstrate that CTZ at 25 μM modulates ΔΨ in PMA treated U1 cells.
Changes in ΔΨ is a distinctive feature of the early stages of apoptosis. Therefore, we assessed levels of cell survival in PMA treated U1 cells and PMA plus 25 μM CTZ treated cells, processed by flow cytometry following propidium iodide (PI) staining. CTZ induced apoptosis by 3-fold in PMA treated cells in comparison to PMA alone treated cells (Fig. 4A).
Figure 4.
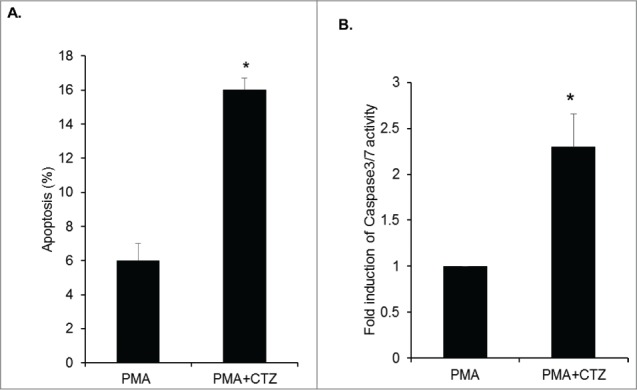
Induction of apoptosis by CTZ in U1 cells and activation of Caspase 3/7. (A) Propidium iodide staining shows an increase in the percentage of apoptotic cells in PMA differentiated U1 cells and PMA differentiated U1 cells treated with CTZ (25μM). * indicates p value <0.02 in comparison to PMA alone. (B) Fold induction in caspase 3/7 activity in PMA differentiated U1 cells and PMA differentiated U1 cells treated with 25μM CTZ. The relative caspase3/7 activity was normalized to protein concentration and the fold induction in the control cells was set at 1. * indicates p value <0.02 in comparison to PMA alone.
The cysteine aspartic acid-specific protease (caspase) family members Caspase-3 and -7 play an important role in apoptosis of mammalian cells.28,29 Therefore, we evaluated whether CTZ induced apoptosis following dissociation of HK-1 from mitochondria in U1 cells treated with CTZ using a Caspase-Glo 3/7 assay that utilizes luminogenic caspase-3/7 substrate for measuring caspase activity. Our results demonstrate a 2.3-fold increase in caspase-3/7 activity in CTZ treated cells in comparison to control cells (Fig. 4B).
Role of HIV-1 Vpr in modulation of HK-1 translocation
Having observed increased levels of HK-1 in the mitochondria of HIV-1 infected macrophages we asked the question whether viral protein Vpr modulates subcellular translocation of HK-1. Recently, using SILAC based proteomic analysis we demonstrated that HIV-1 Vpr induces the expression of HK-1 in macrophages.10 In addition, studies have shown that Vpr is essential for HIV-1 infection of macrophages as the virus replicates less efficiently in macrophages deficient in Vpr.25 We assessed the effect of Vpr overexpression on HK-1 levels in the cytoplasmic and mitochondrial fractions of U937 derived macrophages. The purity of the mitochondrial and cytoplasmic fractions was ascertained using COXIV as a mitochondrial and tubulin as a cytoplasmic marker. We first assessed the levels of Vpr expression in U937 cells transduced with Ad-Vpr and then quantified total HK activity in these cells. These studies show that overexpression of Vpr in Ad-Vpr transduced macrophages (Fig. 5A) inhibited total HK activity in whole cell lysates (Fig. 5B). Interestingly, Ad-Vpr significantly increased HK-1 levels in the mitochondrial fraction in comparison to Ad-Null transduced U937 cells (Figs. 5C, D) with a significant decrease in the cytoplasmic fraction (Figs. 5C, E). These observations demonstrate that Vpr positively modulates the translocation of HK-1 from the cytoplasm to the mitochondria. To show the significance of Vpr mediated translocation of HK-1 to the mitochondria we have used CTZ to dissociate HK-1 from the mitochondria in Vpr transduced U937 cells and assessed activation of Caspase 3/7. The results show a 2.2-fold increase in Caspase 3/7 activity in U973-Vpr transduced cells treated with 25 μM of CTZ in comparison to control (U973-Vpr transduced) cells (Fig. 5F).
Figure 5.

HIV-1 Vpr induces translocation of HK-1 to mitochondria. (A) Representative Western blot of Vpr levels in PMA differentiated U937 transduced with Adeno-Null or Adeno-Vpr. (B) Total HK enzyme activity in arbitrary values (OD 340 nm normalized to protein concentration) in PMA differentiated U937 cells transduced with Adeno-Null or Adeno-Vpr. * indicates p value < 0.05. (C) Representative Western blot of cytoplasmic and mitochondrial fractions from PMA differentiated U937 transduced with Adeno-Null or Adeno-Vpr probed for HK-1. Cytochrome c oxidase subunit IV (COXIV) and α-tubulin was as used markers for fraction purity and protein loading of mitochondrial and cytoplasmic fractions, respectively. (D) Densitometric analysis of HK-1 expression levels in the mitochondrial fraction normalized to COXIV levels in Ad-Null and Ad-Vpr, shown as HK-1/COXIV ratio on the y-axis in arbitrary unit (AU). * indicates p value <0.02 in comparison to Ad-Null. (E) Densitometric analysis of HK-1 expression levels in the mitochondrial fraction normalized to α-tubulin levels in Ad-Null and Ad-Vpr shown as HK-1/Tubulin ratio on the y-axis in arbitrary unit (AU). * indicates p value <0.05 in comparison to Ad-Null. (F) Fold induction in caspase 3/7 activity in PMA differentiated U937 cells transduced with Vpr (Vpr) and PMA differentiated U937 cells transduced with Vpr treated with 25 μM CTZ (Vpr+CTZ). The relative caspase 3/7 activity was normalized to protein concentration and the fold induction in the control cells was set at 1. * indicates p value <0.02 in comparison to PMA alone.
In summary, these observations elucidate the importance of Vpr mediated mitochondrial association of HK-1 in maintaining the integrity of mitochondria since dissociation of mitochondrial attachment of HK-1 with CTZ induced activation of Caspase 3/7 activity in Vpr transduced cells.
Discussion
In comparison to uninfected monocytic cells, persistently infected pro-monocytic cells are less sensitive to apoptotic stimuli.30 HIV-1 infected macrophages are more resistant to apoptosis than other cells infected with HIV-1 and are considered as viral reservoirs.5,31,32 Furthermore, dissemination of virus from these potential reservoirs can promote viral spread to other susceptible target cells.33 The mechanisms responsible for apoptosis resistance in HIV-1 infected macrophages are not well understood. However, it is suggested that macrophages may develop an intrinsic resistance to apoptosis due to differentiation34 or indirectly due to HIV-1 infection35 or as a result of expression of specific viral proteins such as Tat and Nef.36-38 Studies thus far, have demonstrated that induction of macrophage colony-stimulating factor (M-CSF) expression by HIV-1 in macrophages promotes upregulation of anti-apoptotic proteins such as Mcl-1 and Bfl-1.4 Studies have also shown that activation of the stress-induced PI3K/Akt cell survival pathway by HIV-1 infection also protects the infected macrophages from apoptosis.39 Decreased caspase-3 activation is suggested to be a possible mechanism for apoptosis resistance in cells persistently infected with HIV-1.40 Recently, we demonstrated that HIV-1 Vpr induces the expression of HK-1 in macrophages.10 Furthermore, studies have shown that Vpr is essential for HIV-1 infection of macrophages since virus deficient in Vpr is less efficient in replication in macrophages.25
Due to the close relationship between glycolysis and the apoptotic pathway,41 we focused on elucidating whether HIV-1 hijacks the glycolytic pathway to confer a survival advantage in HIV infected macrophages. Hexokinase is a significant regulator of cell death15,22-24 Therefore, modulation of HK expression, activity and subcellular localization in HIV-1 infected cells may induce an anti-apoptotic milieu. Our data demonstrate upregulation of HK-1 expression in HIV-1 infected monocytes and U1 cells induced with PMA with no significant change in the expression of HK-2 protein. However, surprisingly we observed significant decrease in total enzymatic activity of HK following viral infection in PBMCs and after induction of viral replication in chronically infected U1 cells by PMA. These observations are in agreement with a recent study showing decrease in G-6-P levels in U1 cells after induction of viral replication.42 The increase in HK-1 protein expression in HIV-1 infected PBMCs and U1 cells raises the question about the role of this excess HK-1 protein in the infected cells. Studies have demonstrated interaction between HK-1 and VDAC16,43,44 that is mediated by the association of a hydrophobic region of 21 amino acids in the N-termini of both HK-1 and HK-2 with VDAC on the OMM.12,13 This interaction of HK with the mitochondria has been shown to protect cells against cell death.15,22,23 Mechanistic studies show that interaction of HK-1 with mitochondria prevents opening of the mitochondrial permeability transition pore as well as the release of cytochrome c.16 Analysis of the subcellular localization of HK-1 in U1 cells after induction of viral replication with PMA demonstrated an increase in mitochondrial HK-1 level suggesting that viral replication modulates HK-1 association with the mitochondrial membrane of infected macrophages to support mitochondrial health. On the contrary, subcellular localization of HK-1 in U937 cells with PMA did not induce a significant increase in mitochondrial HK-1 level. In this context, it is important to emphasize that PMA, not only induces differentiation of monocytes into macrophages but also independently stimulates viral replication, an effect that can influence the susceptibility to apoptosis.45 These data suggest that apoptosis resistance in the persistently infected U1 cells is also dependent on the magnitude of viral replication.
Earlier studies have shown that for HK to prevent apoptosis, it must localize to mitochondria by binding to VDAC.46 Furthermore, displacement of HK from VDAC by a peptide corresponding to its N terminus15 and dissociation of HK from mitochondria under glucose deprivation also increase apoptosis.47 Our studies also demonstrate the role of Vpr in modulation of HK-1 translocation to mitochondria.
To elucidate the functional significance of mitochondrial HK-1 association and mitochondrial integrity, we used a direct approach to detach HK-1 from the mitochondria while maintaining constant total cellular levels of HK-1. To this end, we used the anti-fungal imidazole derivative CTZ, that dissociates mitochondrial bound HK15,22,26 to demonstrate a significant decrease in ΔΨ using the membrane permeable dye, JC-1 and activation of caspase 3/7 activity in PMA differentiated U1 cells and in Vpr-transduced U937 cells.
This study provides novel evidence that resistance to apoptosis in persistently HIV-1-infected macrophages involves direct modulation of the mitochondrial apoptosis pathway by regulating HK-1 interaction with the mitochondria (Fig. 6) since CTZ accentuates apoptosis by dissociation of mitochondria bound HK-1. Further experiments are needed to elucidate the mechanism by which the HIV-1 and Vpr modulate HK-1 translocation to the mitochondria and control the execution of apoptosis.
Figure 6.
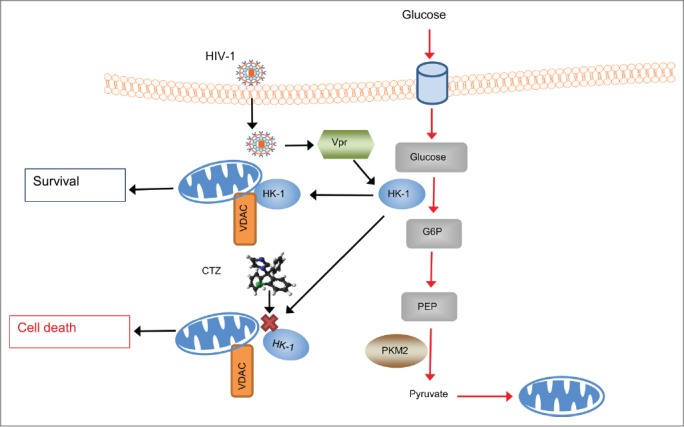
Schematic representation of the anti-apoptotic role of HK-1 in HIV-1 infected macrophages. This mechanism involves up-regulation of HK-1 expression by HIV-1 in a Vpr- dependent fashion that results in increased translocation of HK-1 to the outer mitochondrial membrane (OMM) and interaction with VDAC to promote an anti-apoptotic environment and thereby enhance survival of infected macrophages. However, dissociation of the association of HK-1 to the OMM and interaction with VDAC using a pharmacological inhibitor, clotrimazole (CTZ) indicated by red cross (X) releases HK-1 from mitochondria and induces mitochondrial membrane depolarization and caspase-3/7 mediated apoptosis or cell death.
Materials and Methods
Isolation of PBMCs and HIV-1 infection
Buffy coats were provided by the Comprehensive NeuroAIDS center (CNAC) under the approval of the Temple University Institutional Review Board (IRB). No informed consent was obtained as the study was classified as exempt (category 4) by the IRB.
PBMCs were isolated from buffy coat using the standard ficoll gradient centrifugation method and resuspended in RPMI media containing 10% FBS and 10 mg/ml gentamicin (Life Technologies). Isolated PBMCs were induced with phytohemagglutinin (PHA) (Life Technologies, Cat# 10576–015) at 5 μg/ml for 48 h. PBMCs were then infected with 10000 TCID50/106 cells (MOI ∼0.01) of HIV-1 SF16248 overnight in a 37ºC incubator with 5% CO2. The next day, cells were washed thrice with 1 X PBS and grown in RPMI media containing 10% FBS and 10 mg/ml gentamicin. Six days post infection with HIV-1, cells were harvested for preparation of cell lysates and an aliquot of supernatants were collected to quantify viral load by p24 ELISA (Advanced Bioscience Laboratories, Cat #5421).
Cell culture
U1 cells that contain 2 copies of non-replicating HIV-1 provirus derived from U937 cells chronically infected with HIV-1 were used a model of macrophage latency.18 U1 and U937 cells were maintained in RPMI medium supplemented with 10% FBS and gentamicin in a 37ºC incubator with 5% CO2.
Induction of HIV-1 replication in U1 cells
HIV-1 replication was induced in the U1 cells by treatment with 10 nM of Phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, Cat #P1585) over-night, and the media was replaced with fresh RPMI supplemented with 10% FBS. To quantify viral load, supernatants from the infected cells were collected 3 d post induction of HIV-1 and p24 levels were assessed using the p24 antigen capture assay ELISA kit (Advanced Bioscience Laboratories).
HK Enzyme activity assay
Total HK activity was measured as previously described.49 Briefly, undifferentiated U1 and U937 cells and PMA differentiated U1 and U937 cells were lysed in 50 mM potassium phosphate, 2 mM dithiothreitol (DTT), 2 mM EDTA, and 20 mM NaF. Protein concentration in the lysates was determined by using BCA reagent (Thermo Scientific). HK activity was determined using 30 μg of cell lysates in 1 ml of 100 mM Tris-HCl, pH 8.0, 0.5 mM EDTA, 10 mM ATP, 10 mM MgCl2, 2 mM glucose, 0.1 mM NADP, and 0.1 U/ml of G6PD (Sigma-Aldrich). HK activity was determined by following the G6P-dependent conversion of NADP to NADPH spectrophotometrically at 340 nm using a Beckman DU640B Spectrophotometer.
Isolation of mitochondria and cytoplasmic fractions
Cytoplasmic and mitochondrial fractionation of cells was conducted following an established protocol.50 In brief, cell pellets were resuspended in 1 ml of mitochondria isolation buffer (250 mM sucrose, 10 mM Tris-HCl pH 7.4, 0.1 mM EGTA) and centrifuged at 1,000 rpm for 10 min to pellet nuclei. The supernatant was centrifuged at 15,000 rpm for 20 min to pellet mitochondria. Protein concentration from cytosolic and mitochondrial fractions was determined using BCA reagent (Thermo Scientific).
Adenoviral transduction
Recombinant adenoviral vector harboring HIV-1 Vpr cDNA from the dual-tropic (CCR5 and CXCR4) strain of HIV-1 89.6 was used for overexpression of Vpr.10 PMA differentiated U937 cells (5 X 106) were transduced with adenoviral stock of Ad-Null (empty vector) or Ad-Vpr at multiplicity of infection (MOI) of 10 pfu (plaque forming units) per cell.
Analysis of effects of Clotrimazole on subcellular localization of HK-1
U1 cells were induced by PMA, and 72 h post-induction, cells were treated with 10 and 25 μM of clotrimazole (CTZ) (Sigma-Aldrich, Cat# C6019) or DMSO (Sigma-Aldrich, Cat# D2650) as vehicle control for 10 h. Cells were harvested by centrifugation and washed with PBS and processed for isolation of mitochondria and cytoplasmic fractions as described in earlier section (Isolation of mitochondria and cytoplasmic fractions).
Western blot
Proteins from cells were isolated in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1% Na-deoxycholic acid and 0.1% SDS) in the presence of HALT phosphatase and protease inhibitor (Thermo Scientific). Protein concentration in lysates was determined using BCA reagent (Thermo Scientific) and stored at −70°C until use. Total cell lysates (50 μg) or cytoplasmic fractions (40 μg) or mitochondrial fractions (20 μg) were separated by SDS-PAGE and electroblotted onto a nitrocellulose membrane (Li-Cor Biotechnology). The membrane was blocked with 5% milk in Tris-buffer saline solution (pH 7.6) containing 0.05% Tween-20 (TBS/T) and probed with antibodies diluted at 1:500 (anti-HK-1, #2804S; HK-2, #2106S; COXIV, #4844S; VDAC1 #4866S; Grb2 #3972S antibodies from Cell Signaling Technologies) and α-Tubulin (Sigma-Aldrich). Membranes were incubated with primary antibodies overnight at 4°C. The membranes were then washed in 1 X TBS/T, and then incubated with appropriate secondary antibodies conjugated with Licor IR dyes for 1 h. Images were captured using Odyssey CLx and analyzed using Image Studio Software (Li-Cor Biotechnology).
Analysis of changes in mitochondrial membrane potential (ΔΨ)
U1 cells were induced by PMA and 72 h post-induction, cells were treated for 10 h with 25 μM of CTZ or DMSO as vehicle control. After treatment, cells were harvested by centrifugation and washed with PBS. Cells were labeled with JC-1 dye (Invitrogen) following the manufacturer's protocol. The percentage of cells containing depolarized mitochondria was determined with the Guava EasyCyte mini system (Guava Technologies) by analyzing the ratio of conversion from red to green fluorescence.
Analysis of apoptosis by propidium iodide staining
PMA differentiated U1 cells (72h post-induction) were treated with 25 μM of CTZ or DMSO (vehicle control) for 10 h. Cells were harvested by centrifugation, washed with PBS and then fixed in 70% ice-cold ethanol for 24 h at −20°C. The cells were then washed with PBS and stained with 10 μg/ml of propidium iodide (Sigma-Aldrich) in PBS containing 250 μg/ml RNase A. Following incubation at 37°C for 30 min in the dark, the labeled cells were analyzed by fluorescence activated cell sorting (FACS). The percentage of cells making up the apoptotic cell population was analyzed with the Guava EasyCyte mini system and Guava CytoSoft cell cycle program (Guava Technologies).
Caspase 3/7 activity
U1 cells were induced by PMA and 72h post induction cells were treated for 10 h with 25 μM of CTZ or DMSO as vehicle control. PMA differentiated U937 cells were transduced with Vpr as described in the section on adenoviral transduction. The cells were then treated for 10 h with 25 μM of CTZ or DMSO as vehicle control. Cells were washed with 1X PBS, and Caspase-3/7 activity was measured using a luminescent caspase-Glo 3/7 assay kit (Promega Corporation) following the manufacturer's instructions. An aliquot of cells was also lysed in parallel in M-PER protein extraction reagent (Thermo Scientific) and the protein concentration in the lysates was quantified using BCA protein reagent (Thermo Scientific). Luminescent signal was measured using a Femtomaster FB12 Luminometer (Zylux Corporation).
Statistical analyses
Data were plotted using Windows Excel 2010 (Microsoft). Significance was determined using Student's t-test. A p value of < 0.05 was considered significant. Plotted data represent the mean ± SD of two or 3 experiments.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: U1/HIV-1 from Dr. Thomas Folk and HIV-1SF162 from Dr. Jay Levy.
Funding
This work was supported in part by an NIH/NIDA grant (5R01DA033213–04) to PKD and an NIH grant (NS043980) to SA. This study utilized services offered by core facilities of the Comprehensive NeuroAIDS Center (CNAC, NIMH grant number P30MH092177) at Temple University School of Medicine. We thank Martyn K. White for critically reading the manuscript.
References
- 1. Locati M, Mantovani A, Sica A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv Immunol. 2013; 120:163-84; PMID:24070384; http://dx.doi.org/ 10.1016/B978-0-12-417028-5.00006-5 [DOI] [PubMed] [Google Scholar]
- 2. Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986; 233:1089-93; PMID:3016903; http://dx.doi.org/ 10.1126/science.3016903 [DOI] [PubMed] [Google Scholar]
- 3. Cosenza MA, Zhao ML, Lee SC. HIV-1 expression protects macrophages and microglia from apoptotic death. Neuropathol Appl Neurobiol. 2004; 30:478-90; PMID:15488024; http://dx.doi.org/ 10.1111/j.1365-2990.2004.00563.x [DOI] [PubMed] [Google Scholar]
- 4. Swingler S, Mann AM, Zhou J, Swingler C, Stevenson M. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog. 2007; 3:1281-90; PMID:17907802; http://dx.doi.org/ 10.1371/journal.ppat.0030134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Le Douce V, Herbein G, Rohr O, Schwartz C. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology. 2010; 7:32; PMID:20380694; http://dx.doi.org/ 10.1186/1742-4690-7-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Busca A, Saxena M, Kumar A. Critical role for antiapoptotic Bcl-xL and Mcl-1 in human macrophage survival and cellular IAP1/2 (cIAP1/2) in resistance to HIV-Vpr-induced apoptosis. J Biol Chem. 2012; 287:15118-133; PMID:22403404; http://dx.doi.org/ 10.1074/jbc.M111.312660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Desplats P, Dumaop W, Smith D, Adame A, Everall I, Letendre S, Ellis R, Cherner M, Grant I, Masliah E. Molecular and pathologic insights from latent HIV-1 infection in the human brain. Neurology. 2013; 80:1415-23; PMID:23486877; http://dx.doi.org/ 10.1212/WNL.0b013e31828c2e9e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lamers SL, Fogel GB, Nolan DJ, McGrath MS, Salemi M. HIV-associated neuropathogenesis: a systems biology perspective for modeling and therapy. Biosystems. 2014; 119:53-61; PMID:24732754; http://dx.doi.org/ 10.1016/j.biosystems.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dahiya S, Irish BP, Nonnemacher MR, Wigdahl B. Genetic variation and HIV-associated neurologic disease. Adv Virus Res. 2013; 87:183-240; PMID:23809924; http://dx.doi.org/ 10.1016/B978-0-12-407698-3.00006-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barrero CA, Datta PK, Sen S, Deshmane S, Amini S, Khalili K, Merali S. HIV-1 Vpr modulates macrophage metabolic pathways: a SILAC-based quantitative analysis. PLoS One. 2013;8:e68376; PMID:23874603; http://dx.doi.org/ 10.1371/journal.pone.0068376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003; 206:2049-57; PMID:12756287; http://dx.doi.org/ 10.1242/jeb.00241 [DOI] [PubMed] [Google Scholar]
- 12. Xie GC, Wilson JE. Rat brain hexokinase: the hydrophobic N-terminus of the mitochondrially bound enzyme is inserted in the lipid bilayer. Arch Biochem Biophys 1988; 267:803-10; PMID:3214181; http://dx.doi.org/ 10.1016/0003-9861(88)90090-2 [DOI] [PubMed] [Google Scholar]
- 13. Sui D, Wilson JE. Structural determinants for the intracellular localization of the isozymes of mammalian hexokinase: intracellular localization of fusion constructs incorporating structural elements from the hexokinase isozymes and the green fluorescent protein. Arch Biochem Biophys 1997; 345:111-25; PMID:9281318; http://dx.doi.org/ 10.1006/abbi.1997.0241 [DOI] [PubMed] [Google Scholar]
- 14. Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie. 2002; 84:153-166; PMID:12022946; http://dx.doi.org/ 10.1016/S0300-9084(02)01375-5 [DOI] [PubMed] [Google Scholar]
- 15. Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002; 277:7610-18; PMID:11751859; http://dx.doi.org/ 10.1074/jbc.M109950200 [DOI] [PubMed] [Google Scholar]
- 16. Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondriamediated apoptotic cell death. Biochem J 2004; 377:347-55; PMID:14561215; http://dx.doi.org/ 10.1042/BJ20031465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cassol E, Alfano M, Biswas P, Poli G. Monocyte-derived macrophages and myeloid cell lines as targets of HIV-1 replication and persistence. J Leukoc Biol. 2006; 80:1018-30; PMID:16946020; http://dx.doi.org/ 10.1189/jlb.0306150 [DOI] [PubMed] [Google Scholar]
- 18. Folks TM, Justement J, Kinter A, Dinarello CA, Fauci AS. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 1987; 238:800-02; PMID:3313729; http://dx.doi.org/ 10.1126/science.3313729 [DOI] [PubMed] [Google Scholar]
- 19. Bristow CL, Wolkowicz R, Trucy M, Franklin A, Di Meo F, Kozlowski MT, Winston R, Arnold RR. NF-kappaB signaling, elastase localization, and phagocytosis differ in HIV-1 permissive and nonpermissive U937 clones. J Immunol. 2008; 180:492-99; PMID:18097051; http://dx.doi.org/ 10.4049/jimmunol.180.1.492 [DOI] [PubMed] [Google Scholar]
- 20. Fernandez Larrosa PN, Croci DO, Riva DA, Bibini M, Luzzi R, Saracco M, Mersich SE, Rabinovich GA, Martinez Peralta L. Apoptosis resistance in HIV-1 persistently-infected cells is independent of active viral replication and involves modulation of the apoptotic mitochondrial pathway. Retrovirology. 2008; 5:19; PMID:18261236; http://dx.doi.org/ 10.1186/1742-4690-5-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Olivares I, Ballester A, Lombardia L, Dominguez O, Lopez-Galindez C. Human immunodeficiency virus type 1 chronic infection is associated with different gene expression in MT-4, H9 and U937 cell lines. Virus Res. 2009; 139:22-31; PMID:19000723; http://dx.doi.org/ 10.1016/j.virusres.2008.09.010 [DOI] [PubMed] [Google Scholar]
- 22. Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004; 16:819-30; PMID:15574336; http://dx.doi.org/ 10.1016/j.molcel.2004.11.014 [DOI] [PubMed] [Google Scholar]
- 23. Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. (2008) Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol. 2008; 28:1007-17; PMID:18039843; http://dx.doi.org/ 10.1128/MCB.00224-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu R, Wyatt E, Chawla K, Tran M, Ghanefar M, Laakso M, Epting CL, Ardehali H. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol Med. 2012; 4:633-46; PMID:22517678; http://dx.doi.org/ 10.1002/emmm.201200240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Connor RI, Chen BK, Choe S, Landau NR. (1995) Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995. 206:935-44; PMID:7531918; http://dx.doi.org/ 10.1006/viro.1995.1016 [DOI] [PubMed] [Google Scholar]
- 26. Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P, Rasola A. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS One. 2008. 19;3(3):e1852; PMID:18350175; http://dx.doi.org/ 10.1371/journal.pone.0001852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 2005; 25:4683-96; http://dx.doi.org/ 10.1038/sj.onc.1209595 [DOI] [PubMed] [Google Scholar]
- 28. Nicholson DW, Thornberry NA. Caspases: Killer proteases. Trends Biochem. Sci. 1997; 22:299-306; PMID:9270303; http://dx.doi.org/ 10.1016/S0968-0004(97)01085-2 [DOI] [PubMed] [Google Scholar]
- 29. Thornberry NA, Lazebnik Y. Caspases: Enemies within. Science 1998; 281:1312-16; PMID:9721091; http://dx.doi.org/ 10.1126/science.281.5381.1312 [DOI] [PubMed] [Google Scholar]
- 30. Pinti M, Biswas P, Troiano L, Nasi M, Ferraresi R, Mussini C, Vecchiet J, Esposito R, Paganelli R, Cossarizza A. Different sensitivity to apoptosis in cells of monocytic or lymphocytic origin chronically infected with human immunodeficiency virus type-1. Experimental Biology and Medicine. 2003; 228:1346-54; PMID:14681550 [DOI] [PubMed] [Google Scholar]
- 31. Lum JJ, Badley AD. Resistance to apoptosis. Mechanism for the development of HIV reservoirs. Curr. HIV Res. 2003; 1:261-74; PMID:15046251; http://dx.doi.org/ 10.2174/1570162033485203 [DOI] [PubMed] [Google Scholar]
- 32. Kumar A, Abbas W, Herbein G. HIV-1 latency in monocytes/macrophages. Viruses. 2014; 6:1837-60; PMID:24759213; http://dx.doi.org/ 10.3390/v6041837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Badley AD, McElhinny JA, Leibson PJ, Lynch DH, Alderson MR, Paya CV. Up-regulation of Fas ligand expression by human immunodeficiency virus in human macrophages mediates apoptosis of uninfected T lymphocytes. J. Virol. 1996; 70:199-206; PMID:8523526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kiener PA, Davis PM, Starling GC, Mehlin C, Klebanoff SJ, Ledbetter JA, Liles WC. (1997) Differential induction of apoptosis by Fas-Fas ligand interactions in human monocytes and macrophages. J. Exp. Med. 1997; 185:1511-16; PMID:9126933; http://dx.doi.org/ 10.1084/jem.185.8.1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guillemard E, Jacquemot C, Aillet F, Schmitt N, Barré-Sinoussi F, Israël N. (2004) Human immunodeficiency virus 1 favors the persistence of infection by activating macrophages through TNF. Virology 2004; 329:371-80; PMID:15518816; http://dx.doi.org/ 10.1016/j.virol.2004.08.030 [DOI] [PubMed] [Google Scholar]
- 36. Zhang M, Li X, Pang X, Ding L, Wood O, Clouse KA, Hewlett I, Dayton AI. Bcl-2 upregulation by HIV-1 Tat during infection of primary human macrophages in culture. J. Biomed. Sci. 2002; 9:133-39; PMID:11914580; http://dx.doi.org/ 10.1007/BF02256024 [DOI] [PubMed] [Google Scholar]
- 37. Olivetta E, Federico M. HIV-1 Nef protects human monocyte-derived macrophages from HIV1-induced apoptosis. Exp. Cell Res. 2006; 312:890-900; PMID:16445909; http://dx.doi.org/ 10.1016/j.yexcr.2005.12.003 [DOI] [PubMed] [Google Scholar]
- 38. Abbas W, Khan KA, Kumar A, Tripathy MK, Dichamp I, Keita M, Mahlknecht U, Rohr O, Herbein G. Blockade of BFA-mediated apoptosis in macrophages by the HIV-1 Nef protein. Cell Death Dis. 2014; 5:e1080; PMID:24556695; http://dx.doi.org/ 10.1038/cddis.2014.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lucas A, Kim Y, Rivera-Pabon O, Chae S, Kim DH, Kim B. Targeting the PI3K/Akt cell survival pathway to induce cell death of HIV-1 infected macrophages with alkylphospholipid compounds. PLoS One. 2010; 5:e13121; PMID:20927348; http://dx.doi.org/ 10.1371/journal.pone.0013121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tanaka Y, Kameoka M, Ota K, Itaya A, Ikuta K, Yoshihara K. Establishment of persistent infection with HIV-1 abrogates the caspase-3-dependent apoptotic signaling pathway in U937 cells. Experimental cell research. 1999; 247:514-24; PMID:10066379; http://dx.doi.org/ 10.1006/excr.1998.4376 [DOI] [PubMed] [Google Scholar]
- 41. King A, Gottlieb E. Glucose metabolism and programmed cell death: an evolutionary and mechanistic perspective. Curr Opin Cell Biol. 2009; 21:885-93; PMID:19850457; http://dx.doi.org/ 10.1016/j.ceb.2009.09.009 [DOI] [PubMed] [Google Scholar]
- 42. Hollenbaugh JA, Munger J, Kim B. Metabolite profiles of human immunodeficiency virus infected CD4+ T cells and macrophages using LC-MS/MS analysis. Virology. 2011; 415:153-59; PMID:21565377; http://dx.doi.org/ 10.1016/j.virol.2011.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Linden M, Gellerfors P, Nelson BD. Pore protein and the hexokinase-binding protein from the outer membrane of rat liver mitochondria are identical. FEBS Lett. 1982; 141:189-92; PMID:6178620; http://dx.doi.org/ 10.1016/0014-5793(82)80044-6 [DOI] [PubMed] [Google Scholar]
- 44. Vyssokikh MY, Brdiczka D. The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim. Pol 2003; 50:389-404; PMID:12833165 [PubMed] [Google Scholar]
- 45. Pennington KN, Taylor JA, Bren GD, Paya CV. IkappaB kinase dependent chronic activation of NF-kappaB is necessary for p21(WAF1/Cip1) inhibition of differentiation-induced apoptosis of monocytes. Mol Cell Biol. 2001; 21:1930-41; PMID:11238929; http://dx.doi.org/ 10.1128/MCB.21.6.1930-1941.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr. 2008; 40:171-82; PMID:18683036; http://dx.doi.org/ 10.1007/s10863-008-9148-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. John S, Weiss JN, Ribalet B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS One 2011; 6:e17674; PMID:21408025; http://dx.doi.org/ 10.1371/journal.pone.0017674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cheng-Mayer C, Levy JA. Distinct biological and serological properties of human.immunodeficiency viruses from the brain. Ann Neurol. 1988; 23:S58-S61; PMID:3258140. [DOI] [PubMed] [Google Scholar]
- 49. Ahmad A, Ahmad S, Schneider BK, Allen CB, Chang LY, White CW. Elevated expression of hexokinase II protects human lung epithelial-like A549 cells against oxidative injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002; 283:L573-84; PMID:12169577 [DOI] [PubMed] [Google Scholar]
- 50. Shroff EH, Snyder CM, Budinger GR, Jain M, Chew TL, Khuon S, Perlman H, Chandel NS. BH3 peptides induce mitochondrial fission and cell death independent of BAX/BAK. PLoS One. 2009; 4:e5646; PMID:19468307; http://dx.doi.org/ 10.1371/journal.pone.0005646 [DOI] [PMC free article] [PubMed] [Google Scholar]