

Abstract
Background
Disruptive mutation in the CHD8 gene is one of the top genetic risk factors in autism spectrum disorders (ASDs). Previous analyses of genome-wide CHD8 occupancy and reduced expression of CHD8 by shRNA knockdown in committed neural cells showed that CHD8 regulates multiple cell processes critical for neural functions, and its targets are enriched with ASD-associated genes.
Methods
To further understand the molecular links between CHD8 functions and ASD, we have applied the CRISPR/Cas9 technology to knockout one copy of CHD8 in induced pluripotent stem cells (iPSCs) to better mimic the loss-of-function status that would exist in the developing human embryo prior to neuronal differentiation. We then carried out transcriptomic and bioinformatic analyses of neural progenitors and neurons derived from the CHD8 mutant iPSCs.
Results
Transcriptome profiling revealed that CHD8 hemizygosity (CHD8+/−) affected the expression of several thousands of genes in neural progenitors and early differentiating neurons. The differentially expressed genes were enriched for functions of neural development, β-catenin/Wnt signaling, extracellular matrix, and skeletal system development. They also exhibited significant overlap with genes previously associated with autism and schizophrenia, as well as the downstream transcriptional targets of multiple genes implicated in autism. Providing important insight into how CHD8 mutations might give rise to macrocephaly, we found that seven of the twelve genes associated with human brain volume or head size by genome-wide association studies (e.g., HGMA2) were dysregulated in CHD8+/− neural progenitors or neurons.
Conclusions
We have established a renewable source of CHD8+/− iPSC lines that would be valuable for investigating the molecular and cellular functions of CHD8. Transcriptomic profiling showed that CHD8 regulates multiple genes implicated in ASD pathogenesis and genes associated with brain volume.
Electronic supplementary material
The online version of this article (doi:10.1186/s13229-015-0048-6) contains supplementary material, which is available to authorized users.
Keywords: Autism, ASD, CHD8, CRISPR/Cas9, Induced pluripotent stem cells, iPSC, Neurodevelopment, Macrocephaly, RNA-seq, Schizophrenia
Background
ASDs are a class of neurodevelopmental disorders characterized by persistent deficits in social communication/interaction and restricted, repetitive patterns of behaviors, interests, or activities (DSM-5) [1]. The genetic risk factors for ASD are heterogeneous, and up to 1 thousand genes are estimated to be involved [2]. Recent whole exome and genome-sequencing projects, focused on the identification of rare de novo mutation in the probands of ASD family “trios” or “quads”, have discovered hundreds of genes with functionally disruptive mutations [3–12], some of which also map to ASD-associated de novo copy number variations (CNVs) [13]. Many of the implicated genes encode proteins involved in synapse formation, transcriptional regulation, and chromatin remodeling [10, 14], indicating a likely convergence of functional pathways, despite genetic heterogeneity.
Chromodomain helicase DNA binding protein 8 (CHD8) emerged as a top ASD-candidate gene from multiple exome-sequencing studies [6, 8], which altogether have analyzed thousands of ASD probands and in some cases their families too [15, 16]. More importantly, disrupting chd8 in zebrafish development has recapitulated multiple features of ASD, including macrocephaly observed in some ASD cases carrying CHD8 mutations [15, 16]. In addition, macrocephaly is often found in ASD caused by disruption of other candidate genes [17].
CHD8 is a ubiquitously expressed member of the CHD family of ATP-dependent chromatin-remodeling factors that play important roles in chromatin dynamics, transcription, and cell survival [18]. Previous studies [19–21] have shown that the CHD8 protein interacts with β-catenin and negatively regulates Wnt signaling, while both β-catenin and Wnt signaling play critical roles in normal brain development and neuropsychiatric disorders [22], including ASD [14]. CHD8 also functionally interacts with p53 [23] and recruits MLL histone methyltransferase complexes to regulate cell cycle genes [24].
To elucidate the roles of CHD8 in neurodevelopment and address the contribution of its disruption to ASD pathogenesis, three groups have recently reported genome-wide CHD8 binding sites and transcriptomic changes upon shRNA-mediated knockdown of CHD8 expression in neural progenitor cells (NPCs) [25], neural stem cells (NSCs) [26], and SK-N-SH neuroblastoma cells [27]. The results showed that CHD8 binds to thousands of genes, largely biased to promoters, in NPCs, NSCs, and developing mammalian brains. Reduced expression of CHD8 resulted in the potential disruption of gene networks involved in neurodevelopment, which contained many ASD-risk genes [15, 25–27]. The transcriptional targets of CHD8 have also been studied in non-neural systems, as it is also involved in cell cycle regulation, Wnt signaling, and several forms of cancers [19, 21, 24, 28, 29].
As ASD is a developmental disorder with symptoms emerging in early childhood and the CHD8 causal mutations in patients are germline, it is important to establish cell models that can mimic the persistent loss of CHD8 function in the developing embryos prior to and during neuronal differentiation and brain development. Therefore, we applied CRISPR/Cas9 technology [30] to generate CHD8+/− iPSCs by knocking out one copy of the gene in an iPSC line derived from a healthy male subject. We then differentiated both the wild-type (WT) and the CHD8+/− iPSCs to NPCs and subsequently neurons and performed comparative transcriptomic analysis (RNA-seq). Our approach has several advantages: precisely targeted changes at the DNA level, persistent reduction of CHD8 expression, no introduction of extra genetic materials (e.g., virus vector) to the cells, and greater flexibility in the types of differentiated cells that can be generated.
We found that heterozygous CHD8 knockout (KO) disrupted the expression of many genes involved in extracellular matrix formation, neuronal differentiation, and skeletal system development. Interestingly, CHD8-regulated genes were enriched with ASD-risk genes, schizophrenia-risk genes, and genes implicated in regulating head size or brain volume. Furthermore, we found that CHD8-regulated genes significantly overlapped with the downstream targets of several critical genes (TCF4, EHMT1, SATB2, and NRXN1) that have been associated with ASD or other neuropsychiatric disorders. Taken together, our results not only shed light on the molecular roles of CHD8 in neurodevelopment, but also provide evidence of potential convergence of cellular pathways that could be disrupted by mutations of distinct ASD-risk genes.
Methods
Development of iPSCs from skin fibroblasts
We have been developing iPSCs from controls and patients with 22q11.2 deletion and diagnosis of a psychotic disorder [31]. The study and consent forms were approved by the Institutional Review Board of Albert Einstein College of Medicine . Consent was obtained by a skilled member of the research team who had received prior human subject training. One of the healthy male control (without 22q11.2 del too) was used in the current study. Exome sequencing was performed on DNA extracted from white blood cells of this subject to detect coding variants prior to generating the CHD8 KO. We used GATK [32] for variant calling and ANNOVAR [33] for variant annotation.
The iPSC line used in this study was generated from fibroblasts obtained from a skin biopsy performed by a board-certified dermatologist. The procedures for growing fibroblasts and iPSC reprogramming are detailed in Additional file 1. Pluripotency was confirmed by immunocytochemistry using antibodies (Ab) against Tra-1-60, Tra-1-81, SSEA3, and SSEA4, which are expressed in pluripotent stem cells. In addition, the capacity to differentiate into all three germ layers was established by in vitro assays, as previously described (see Additional file 1 for details) [34, 35].
Design of the CHD8 single guide RNA sequences
Single guide RNA (sgRNA) sequences targeting the region adjacent to the Ser62 codon of CHD8 were picked using the online CRISPR design tool [36] from the Zhang lab at the Broad Institute, and the two selected sgRNAs were predicted to have very low probability off-target sites. The sgRNA sequences were then cloned into the pSpCas9 (BB)-2A-Puro (PX459) vector (a gift from Dr. Feng Zhang, Addgene plasmid # 48139) [30].
CRISPR/Cas9-mediated CHD8 knockout
Human iPSCs were cultured and fed daily in mTeSR1 (Stem Cell technologies) on Matrigel (BD)-coated plates at 37 °C/5 % CO2/85 % in a humidified incubator. Cells were maintained in log phase growth, and differentiated cells were manually removed. iPSCs were exposed to 10-μM ROCK Inhibitor for ~4 h to improve cell survival during nucleofection. After 4 h, growth medium was aspirated, and the cells were rinsed with DMEM/F12. iPSCs were dissociated into single cells using accutase and harvested. Nucleofection was performed using the Amaxa-4D Nucleofector Basic Protocol for Human Stem Cells (Lonza) according to the manufacturer’s instructions. Briefly, 8 × 105 cells and 5 μg of the CRISPR/Cas9 plasmids with either sgRNA1 or sgRNA2 were nucleofected using the P3 Primary Cell 4D-Nucleofector X Kit L with program CA-137. Cells were resuspended in mTeSR1 + 10-μM ROCK Inhibitor and placed in one well of a 6-well Matrigel-coated plate. The following day, cells were fed with fresh mTeSR1, and were subsequently fed with fresh medium every day. On days 4–14, cells were exposed to 0.5 μg/ml puromycin for 6 h. Puromycin-resistant colonies were picked and expanded in mTeSR1 without further puromycin treatment.
Characterizing CHD8 knockout lines
“TA” cloning was used to identify the knockout alleles. A 479-bp PCR amplicon flanking the CRSPR/Cas9-targeted sites was generated using the primers 5’-CTGTAAGACAGGTTGGGCTG-3’ and 5’-CTTGTTTCTTGCCTCTATACTTGA-3’. The PCR product was purified and ligated into pCR™2.1 using a TA Cloning Kit developed by Life Technologies following the manufacturer’s protocol. Recombinant plasmids were introduced into competent E. coli and selected in ampicillin. Plasmid DNA was extracted and sequenced across the insert using one of the PCR primers.
Western blotting confirmed that the CHD8+/− lines expressed lower levels of CHD8 protein. Specifically, cell lysates from NPCs differentiated from WT, and KO iPSCs were separated by SDS PAGE, transferred to PVDF membranes, and then blotted with anti-CHD8 antibody (Bethyl Cat #A301-224A). Anti-actin antibody (BD Biosciences, Cat # 612656) was used for loading control.
Neuronal differentiation
Neurons were generated from iPSC-derived NPCs as described by Marchetto et al. with slight modifications [34, 37]. A detailed description of the protocol can be found in Additional file 1. Essentially, the protocol leads to a mixed population of glutamatergic and GABAergic neurons, from which RNA was extracted and sent for sequencing.
RNA-seq analysis
We obtained 101-bp paired-end RNA-seq reads from Illumina HiSeq 2500. RNA-seq reads were aligned to the human genome (hg19) using Tophat (version 2.0.8) [38]. The number of RNA-seq fragments mapped to each gene was determined for genes in the GENCODE database (v18) [39]. Exonic/intronic/intergenic rates were calculated by CollectRnaSeqMetrics in Picard [40]. Cufflink (version 2.2.1) [41] was used to generate the gene-expression values as FPKMs (fragments per kilobase of exon per million fragments mapped). We restricted our analysis to 12,843 protein-coding genes with average FPKM >1 across all four samples. DESeq2 [42] was used to determine differentially expressed genes (DEGs) in NPCs and neurons. The list of significantly DEGs was defined at false discovery rate (FDR) < 0.05. DAVID [43, 44] was used for Gene Ontology (GO) analysis with 12,843 expressed protein-coding genes as background. Ingenuity pathway analysis (IPA) [45] was used for canonical pathway analysis and disease association, with the ingenuity knowledge base (genes only) as background. Toppgene [46] was used for human phenotype ontology analysis, and the whole genome was used as background. The RNA-seq data have been deposited in the Gene Expression Omnibus (GEO; accession # GSE71594).
To find neurodevelopment genes specifically affected by CHD8+/−, we added an interaction design in DESeq2 (option: ~celltype + genotype + celltype:genotype) to specifically model the interactions between development status (NPCs or neurons) and CHD8 status (WT or KO).
Validating targeted deletions and assessing off-targets using RNA-seq reads
First, we examined if CHD8 was targeted and edited precisely according to our CRISPR sgRNA design. A 2-bp deletion in chr14:21899785 (hg19) and a 10-bp deletion in chr14:21899722 (hg19) were identified in a proportion of RNA-seq reads that mapped to targeted regions of the two CRISPR sgRNAs in CHD8+/− samples (KO1 and KO2, respectively). This was confirmed by DNA sequencing. The two short indels were not found in any of the WT samples. We also called short indels (supported by at least five RNA-seq reads) from RNA-seq reads by samtools [47], but we did not detect any additional indels that were present in CHD8+/− samples but not in the WT controls.
Quantitative real-time PCR (qPCR)
qPCR was carried out on reverse transcribed PCR using the 2−ΔΔCt method as previously described [48, 49]. A detailed description and the primers used for this analysis can be found in Additional file 1.
Definition of CHD8-binding genes
ChIP-seq peaks of CHD8-binding sites in NPCs were from Sugathan’s report [25]. Only peaks replicated by all three antibodies were used. Genes with at least one peak from 2 kb upstream of the transcription start sites to the transcription terminus were defined as CHD8-binding genes.
Interaction network analysis
DEGs in NPCs with CHD8-binding were imported into the STRING database v10 [50] to construct protein-protein interaction networks. We retained any interaction (i.e., edge) from experiments and databases that had a median confidence ≥0.4.
For detecting converged networks of multiple ASD-risk genes, we first collected DEG lists from our CHD8+/− NPCs, TCF4 knockdown, EHMT1 knockdown, MBD5 knockdown, and SATB2 knockdown, respectively [51, 52], and then imported genes shared by at least two lists into the STRING database to construct gene networks. GO enrichment was calculated by “Enrichment” function in STRING. Networks were visualized using Cytoscape [53]. The same approach was also applied to DEGs from CHD8 KO, ZNF804A KD, and NRXN1 KD neurons.
Upstream regulator prediction
IPA was used to predict upstream regulators for 841 DEGs without CHD8 binding in NPCs. In this analysis, the p value from IPA measures the significance in overlap between query genes and pre-defined sets of genes that are regulated by a specific regulator, using the Fisher test. At the end, we used p < 0.05 to select upstream regulators that (a) regulated at least five non CHD8-bound DEGs and, themselves, were (b) in our NPC DEG list.
ASD/schizophrenia-risk gene sets
The first ASD gene set was obtained from the SFARI gene-scoring module [54], using genes scored as high confidence, to minimal evidence and syndromic. The second ASD gene set was from the AutismKB [55] core dataset, which includes syndromic autism related genes and non-syndromic-related genes, designated as high confidence. High-confidence and probable ASD genes in Willsey’s paper [56] were used as the third set (“Willsey_ASD”). Genes predicted from whole exome-sequencing and co-expression network [57] were used as another set (“Liu_ASD”). The other two lists were derived from massive whole exome sequencing: one (“Iossifov_ASD”) focused on de novo mutations [11] and the other (“DeRubeis_ASD”) combined de novo and inherited mutations to develop a high-confidence list (FDR < 0.1) [10]. Two schizophrenia gene lists were from the SZgene database [58] and a recent GWAS report [59].
Identification of common GO terms for DEG lists from different CHD8 studies
DEGs were determined by the following criteria from data in four previous studies. For the study by Cotney et al. [26], we selected genes with logFC > 0.1 and log counts per million (logCPM) between 2 and 10 to meet the Poisson assumption, as described by the original authors. However, we repeated differential expression analysis with a less stringent FDR cutoff, using the Benjamini-Hotchberg method instead of Bonferroni to adjust p values for significance. For the study by Sugathan et al. [25], we used ComBat in the sva package [60] to adjust batch effects, followed by differential expression analysis with DESeq2 [42]. DEGs were selected by p value < 0.05; 96 % of DEGs in Sugathan’s list were in our reanalysis list. For Wilkinson et al’s study [27], we used the DEG list provided by the authors.
Enriched GO terms for each of the five DEG lists were first determined by ClueGO [61] (p value < 0.05). Subsequently, GO terms shared by ≥3 DEG lists were considered as common and used for subsequent analysis of function overlap between the five CHD8+/− and knockdown studies. The relationships among the selected GO terms were based on their shared genes, which was measured using kappa statistics [62]. Two GO terms were connected by an edge if they had a kappa score >0.4. ClueGO relies on term similarity to define functional groups of multiple terms. In our analysis, we set initial group size to 5 (default value, 2) and percentage for group merge (default values, 50 %) to 80 % to obtain a summary of less redundant functional clusters of common GO terms. Since a GO term can be included in several functional groups, we assigned each term only to the functional groups in which it had the most significant group p values, meaning that this term had the most similar genetic component in this group. Subsequently, terms of the same groups formed a closed circle. In addition to edges connecting terms to show their relationships, we also added edges between terms and studies to reveal enrichment of specific GO terms among individual DEG lists.
Statistics
To test if DEGs were significantly overlapped/enriched with a specific gene set, 12,843 expressed genes in our samples were used as background (of all genes) for the Fisher exact test. Statistics tests were conducted in R [63] and multiple test correction was applied unless specified otherwise.
Results
Characterization of CHD8 knockout iPSC lines
Several functional disruptive mutations, including premature stop codons and frameshift mutations, have been detected in CHD8 in ASD probands [15, 16]. We designed two separate CRISPR sgRNA sequences to target the N-terminal of CHD8 protein to generate truncated mutations (Fig. 1a). iPSCs derived from a healthy male subject were transduced with CRISPR/Cas9 vectors containing each of the two target sequences. After screening, two clones, one with a 2-bp (KO1) and the other with a 10-bp (KO2) heterozygous deletion were found; the other allele was intact in both (Fig. 1b). Both genomic DNA sequencing and Western blotting analysis (Fig. 1b, c) confirmed the heterozygous knockout status for both clones. The CHD8+/− iPSC lines were used to generate NPCs and early differentiating neurons for RNA-seq analysis, together with samples prepared from the parental WT clones, for a total of eight samples (two biological replicates of WT and CHD8+/− at two neurodevelopmental stages).
Fig. 1.
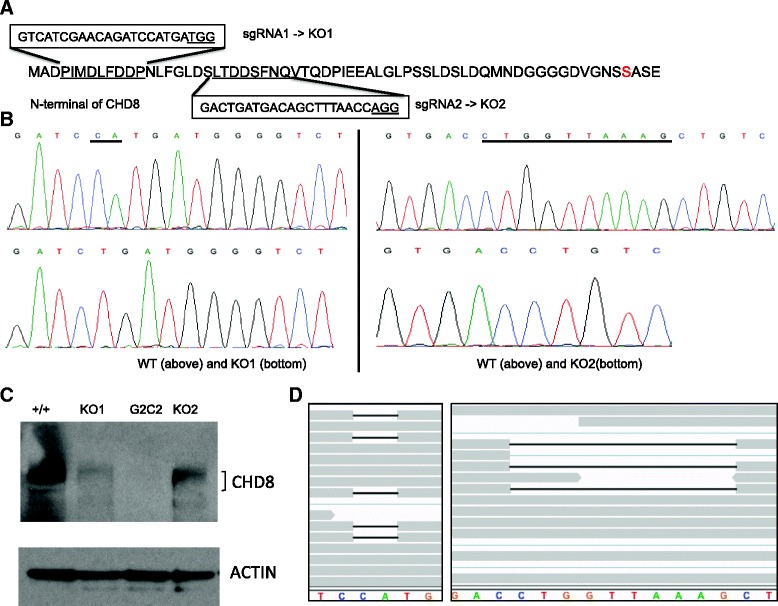
Generation and characterization of CHD8 +/− lines. a Design of CRISPR guide sgRNA sequences targeting the N-terminal end of CHD8. The red marks Ser62 where premature stop codon mutations were uncovered from whole exome-sequencing analysis of ASD individuals [16]. b DNA-sequencing analysis of CHD8 +/− iPSC clones. Knockout alleles were identified by cloning of PCR products and Sanger sequencing. c Western blot analysis of CHD8 +/− NPCs. CHD8-specific antibodies were used to detect CHD8 protein in NPC lysates. A clone with homozygous CHD8 knockout (G2C2) was also analyzed, but this clone could not differentiate into neurons appropriately and thus was not included in our RNA-seq analysis. d Validation of 2-bp (KO1, left) and 10-bp (KO2, right) deletion by RNA-seq reads. A screen shot of RNA-seq reads mapped to the CRISPR targeting regions, with gap showing deletion
Genes with altered expression in CHD8+/− are involved in neurodevelopment
During RNA-seq analysis, we obtained on average ~28 million read pairs per sample (Additional file 2: Table S1). Examination of the RNA-seq reads mapped to the CHD8 exons confirmed the 2-bp and 10-bp deletion (Fig. 1d), indicating that both the WT and KO CHD8 copies were expressed in NPCs and neurons, with fewer reads from the KO copy than the WT. Importantly, our indel analysis of the RNA-seq reads detected no off-target sites at coding regions (see Methods).
Differential expression analysis identified 1248 and 3289 genes that changed expression in NPCs and neurons, respectively (FDR < 0.05, Fig. 2a, Additional file 3: Table S2). Note that CHD8 showed no significant expression change at the transcript level (p = 0.85 in NPCs, p = 0.27 in neurons, Additional file 3: Table S2). The fact that many more DEGs were detected in neurons than NPCs indicates that persistent CHD8 hemizygosity could have continuous and amplified effects in neurodevelopment, i.e., genes with altered expression in NPCs would directly affect the expression of additional sets of genes in the differentiating neurons. Seven DEGs, including SMARCA2, WNT7A, HMGA2, TESC, TCF4, TGFβ3, and DDR2, which are involved in transcription regulation, cell division, and WNT-β-catenin signal pathways, or related to head size or brain volume (see below), were selected for qRT-PCR analysis, and their differential expression in CHD8+/− neurons was confirmed (Fig. 2b). Of them, SMARCA2, HMGA2, and TCF4 were potentially direct targets as they were bound by CHD8 [25].
Fig. 2.
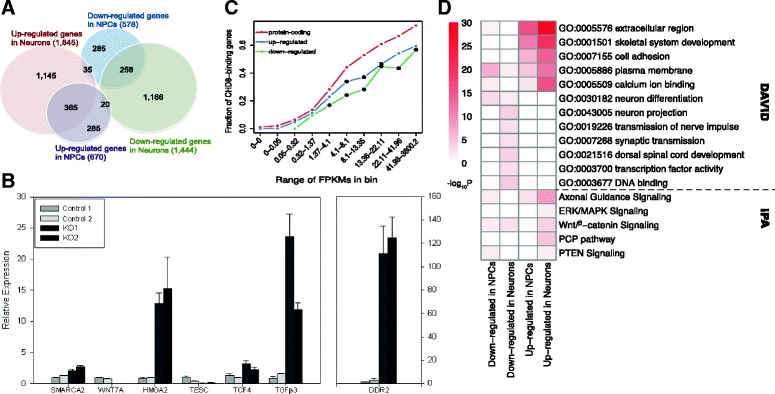
Functional annotation of DEGs and their relationship to CHD8-binding. a Venn diagram of DEGs from CHD8 +/− in iPSC-derived NPCs and neurons. b qRT-PCR validation of seven DEGs, using β2-microglobulin (β2M) as a reference gene to calculate relative expression levels. The qPCR was carried out on neurons derived from two control samples and the two CHD8 +/− clones (the same samples used for the RNA-seq). Each was normalized against another control sample. Samples were analyzed in triplicate. The graph shows the fold change for each sample relative to the neutral control, error bars show the +/− standard deviation. The fold changes were highly significant: SMARCA2, p = 9.7E-7; WNT7A, p = 6.1E-9; HMGA2, p = 4.4E-6; TESC, p = 0.003; TCF4, p = 4.4E-5; TGFB3, p = 5.5E-6; DDR2, p = 6.2E-8 (two-tailed Student’s t test). c Percentage of CHD8-binding genes in all protein-coding, upregulated, and downregulated genes in NPCs, ranked by their expression values (FPKMs) in WT NPCs. Asterisks (“*”) mark the groups of genes showing a significant difference in the proportions of genes with CHD8 binding by comparing DEGs with all protein-coding genes (binomial test, two-tailed, p < 0.05). d Representative-enriched GO terms (top) and canonical pathways (bottom) among DEGs, as reported by DAVID and IPA, respectively. The red color in each cell corresponds to the −log10(p value), corrected by Benjamini-Hotchberg method (color scale on the left and only terms with p < 0.05 were shown)
Previous studies have reported that CHD8 bound to thousands of genomic regions (i.e., peaks), especially to gene promoters in NPCs or NSCs [25, 26]. To explore the relationship between DNA binding and gene regulation, we integrated CHD8 binding in control NPCs [25] with our expression data. Based on the average gene expression of the two WT NPC samples, we separated genes into 10 bins of equal size (1284 genes per bin). Consistent with the results from Sugathan et al. [25], CHD8 binding was observed more frequently at more highly expressed genes (Fig. 2c). To our surprise, we found that the percentages of CHD8 binding genes among the DEGs were significantly lower than the expectation from genome-wide binding (Fig. 2c). Nevertheless, this finding is consistent with previous reports that only a small percentage of CHD8-bound genes displayed differential expression upon CHD8 knockdown [25]. This result indicates that CHD8 directly regulates a limited number of genes and that the majority of gene-expression changes due to CHD8 knockout are indirectly regulated targets.
GO analysis using DAVID revealed that upregulated genes in CHD8+/− NPCs and neurons were enriched with similar GO terms, including “extracellular region,” “skeletal system development,” and “cell adhesion.” On the contrary, for downregulated genes, except for the GO term “neuron differentiation” that was enriched in both NPCs and neurons, “neuron projection,” “synaptic transmission,” and “transcription factor activity” were only enriched in neurons (Fig 2d, see full list in Additional file 4: Table S3). In addition, using IPA, we found that DEGs were highly enriched in “axonal guidance signaling,” “WNT-β-catenin signal,” and “PTEN signaling” (Fig. 2d, full list in Additional file 5: Table S4). These results indicate that heterozygous CHD8 mutations may disrupt multiple processes of neurodevelopment and neural functions in ASD. It should be noted that, in addition to nervous system development, the DEGs were also enriched for cancers, gastrointestinal disease, and cardiovascular system development, based on the IPA analysis (Additional file 5: Table S4).
As genes downregulated in both CHD8+/− NPCs and neurons were enriched for neuronal differentiation, we wondered how heterozygous CHD8 KO might affect gene regulation during the transition from NPCs to differentiating neurons. To address this, we utilized a two-factor linear model implemented in DESeq2 to search for genes whose transitional expression from NPCs to neurons were affected by CHD8 reduction (i.e., interaction between neuronal differentiation and CHD8 status; see Methods). This analysis identified 1098 genes. Among them, 360 also showed a significant differential expression between NPCs and neurons in WT but not in the comparison of CHD8+/− samples. Of these 360 genes, 207 genes were expressed at a higher level in WT neurons (vs. WT NPCs). GO analysis revealed that these genes were enriched in “neurological system process,” especially in “transmission of nerve impulse” and “synaptic transmission” (Additional file 2: Figure S1A). The 153 genes with higher expression in WT NPCs (vs. WT neurons) were enriched for “cell junction” and “cell adhesion” (Additional file 2: Figure S1B). These results indicate that normal synapse formation and function could be disrupted during CHD8+/− neuron differentiation.
CHD8 direct targets vs. indirect targets
To better understand the regulatory roles of CHD8, we further characterized the 407 DEGs in NPCs with CHD8 binding (i.e., direct targets) and used STRING to define their function interactions, which include direct protein-protein interactions and indirect functional associations. Of the 407 genes, 140 were included in the resulted STRING network, and interestingly, those genes could be grouped into several function clusters: cell cycle, cytoskeleton, cell adhesion, chromatin factor, ribonucleoprotein complex, and GTPase genes (Fig. 3a), revealing major pathways that could be directly regulated by CHD8 in NPCs. We did not perform the same analysis for neuron DEGs because no CHD8-binding data was available for human neurons.
Fig. 3.
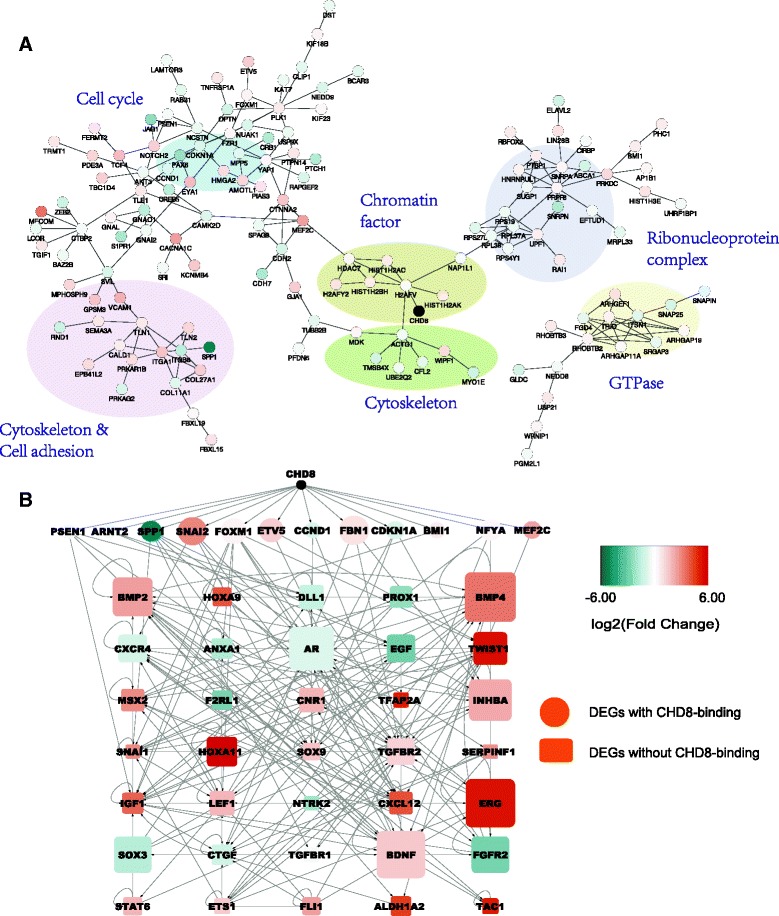
Interaction network of CHD8 direct targets and a putative CHD8 regulatory network in NPCs. a Functional interaction network generated by the STRING database for CHD8 direct targets, with nodes representing genes and edges representing interactions. Colors of the nodes indicate expression changes. Disconnected genes were not shown. b A putative regulatory network connecting CHD8 to a set of upstream regulators, which in turn could regulate the expression of many genes indirectly targeted by CHD8. The edges represent regulatory relationship predicted by the IPA for upstream regulators that were bound (ellipse) or not bound (rectangle) by CHD8. Colors of the nodes indicate expression changes. Sizes of the nodes show the –log10 (p value) from IPA. Arrows start from an upstream regulator to its targets. Note that only differentially expressed upstream regulators were included in the network
As DEGs were statistically depleted of CHD8 binding, we wondered how then those 841 NPC DEGs without CHD8 binding (i.e., indirect targets) could be affected by reduced CHD8 expression. To address this, we applied IPA software to predict the upstream regulators of these CHD8 indirect targets. The results showed that 12 CHD8 direct targets could serve as upstream regulators of 95 of the 841 CHD8 indirect targets (Fig. 3b), including ASD-risk genes MEF2 [64] and ARNT2 [65]. This analysis also revealed that some DEGs could be regulated by 35 DEGs that were, themselves, indirect targets. Together, these 47 upstream regulators formed a complex regulatory network (Fig. 3b), mediating key cellular pathways (e.g., BMP and TGFβ) that eventually may lead to expression changes for thousands of genes when CHD8 expression is disrupted, an interesting finding to be further investigated.
CHD8-regulated genes are overall longer than non-DEGs
We noticed that some extremely long genes were dysregulated in either NPCs or neurons, like LSAMP (2187 kb), PCDH15 (1825 kb), RBFOX1 (1694 kb), and NRXN3 (1622 kb). In addition, many high-confidence ASD-candidate genes are exceptionally long [66]. At the genome-wide level, we found that DEGs in both NPCs and neurons were significantly longer than the non-DEGs (Fig. 4). The length difference between DEGs and non-DEGs was not detected in a separate study in which we compared the iPSC-derived neurons from schizophrenia patients with controls (manuscript submitted; data available in the GEO: GSE46562); the mean lengths were 77 kb for DEGs and 73 kb for non-DEGs (p value = 0.51, Student’s t test). This indicates that our observation is not simply a result of analyzing expression data in the neural induction system we used. This difference was detected for genes with CHD8-binding (Fig. 4a) or without CHD8-binding (Fig. 4b), suggesting that other transcription regulators may cooperate with CHD8 to regulate the expression of long genes.
Fig. 4.
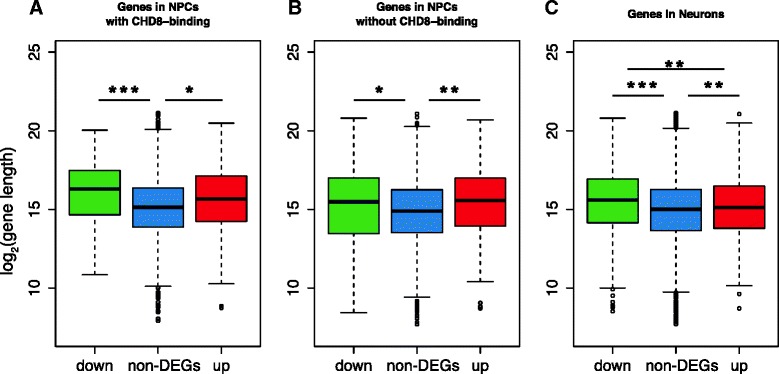
Difference in gene length between DEGs and non-DEGs. Plotted here are length distributions of genes with CHD8 binding in NPCs (a), without CHD8 binding in NPCs (b), and all genes in neurons (c). In NPCs, mean of gene lengths is 117 kb for DEG and 76 kb for non-DEG; in neurons, mean of gene lengths is 98 kb for DEGs and 74 kb for non-DEGs, whereas gene lengths were defined as the distances from transcription start site to termination site in Gencode v18. *p < 0.01, **p < 1e-5, ***p < 1e-9, t test, two-tailed
DEGs are enriched for genes associated with human head size/brain volume
Macrocephaly is overrepresented in ASD patients and a defined feature of some syndromic forms of ASD, including those carrying loss-of-function CHD8 mutations [15, 16]. In addition, chd8 disruption in zebrafish resulted in increased embryonic head size [15, 25]. We thus used Toppgene to predict the potential phenotypes that could result from the dysregulation of CHD8 targets. For downregulated genes in CHD8+/− neurons, “abnormality of skull size” was one of the most significant human phenotypes (Fig. 5a), so were neurodevelopmental abnormality, intellectual disability, and abnormality of brain morphology, all consistent with clinical phenotypes seen in patients with CHD8-disruptive mutations [15]. For the other gene groups, i.e., up- or downregulated genes in CHD8+/− NPCs or upregulated genes in CHD8+/− neurons, either no human phenotypes were significantly enriched, or enriched phenotypes showed no obvious relation to brain or head development (Additional file 6: Table S5). It would be interesting to further study how this difference detected for the DEGs of CHD8+/− NPCs and neurons may be related to changing CHD8 roles at different stages of brain development.
Fig. 5.
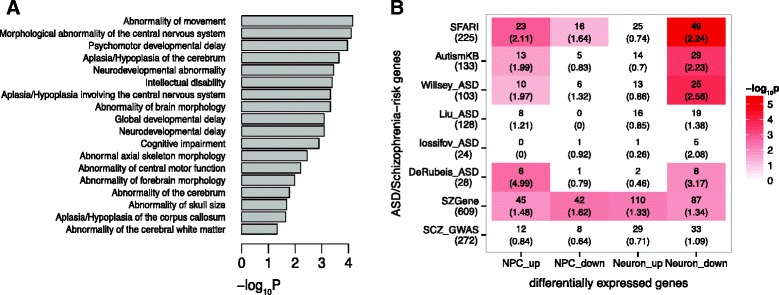
Enrichment of DEGs for ASD/schizophrenia-risk genes and genes associated with specific human phenotypes. a Enriched human phenotype from the Toppgene analysis for downregulated genes in neurons. p values were corrected by the Bonferroni method. b Enrichment of DEGs for ASD- and schizophrenia-risk genes. ASD-risk genes were collected from the SFARI database [54], AutismKB core dataset [55], and four other sets based on whole exome-sequencing and gene-expression network (Willsey_ASD [56], Liu_ASD [57], Iossifov_ASD [11] and DeRubeis_ASD [10]). The number of expressed genes in each set was in left parentheses. Schizophrenia-risk genes were obtained from either the SZGene database [58] or a recent GWAS report [59]. The red color in each cell corresponds to the −log10 (p value) for enrichment (Fisher’s exact test, one-tailed), as shown in the color scale on the right (shown only if p < 0.05). The first and the second number (in parenthesis) within each cell are the number of overlapping genes and the odds ratio of overlap, respectively
Next, we compared the lists of DEGs in CHD8+/− NPCs and neurons with genes previously associated with brain size or volumes of specific brain regions. Recent GWASs revealed a small number of common variants associated with the volumes of different human brain regions or head sizes [67–69]. Remarkably, of the only 12 genes linked to the statistically significant GWAS variants, 7 were differentially expressed in CHD8+/− neurons, which was significantly higher than expected (odds ratio (OR) = 4.07, p = 0.016, Fisher’s exact test, one-tailed; Table 1). HMGA2, a high-confident candidate linked to adult intracranial volume [68] and infant head circumference [69], was a CHD8 direct target (with CHD8 binding to its promoter [25, 26]), and it was upregulated in both CHD8+/− NPCs and neurons. Interestingly, HMGA2 encodes a chromatin-associated regulator important for stem cell renewal. How CHD8 and HMGA2 interact and co-regulate gene-expression and cell cycles warrants further study. Another DEG associated with caudate volume is FAT3, which codes for an atypical cadherin previously shown to affect dendritic pruning [70], a known defect in ASD. Interestingly, by analyzing the spatiotemporal transcriptional data across developing brains available in the Brainspan project [71], we found that the expression of CHD8 was significantly and positively correlated with FAT3 during brain development (Additional file 2: Figure S2), suggesting that CHD8 is a positive regulator of FAT3, consistent with the downregulation of FAT3 in CHD8+/−. MAPT, another top candidate associated with infant head circumference [69], was downregulated in CHD8+/− neuron (nominal p value = 0.015).
Table 1.
Expression changes of genes associated with brain volume or head size from GWAS
| Ref. | Trait | SNP | Associated gene | Coordinate | CHD8-binding in NPCs | CHD8 +/− NPC | CHD8 +/− neuron | ||
|---|---|---|---|---|---|---|---|---|---|
| log2(FC) | q value | log2(FC) | q value | ||||||
| [68] | Hippocampal volume | rs7294919 | TESC | chr12:117476728-117537284 | N | 0.42 | 0.89 | −2.78 | 0.0020 |
| [68] | Total brain volume | rs10494373 | DDR2 | chr1:162601163-162750237 | N | 4.12 | 1.68E-31 | 5.70 | 2.82E-13 |
| [68] | Intracranial volume | rs10784502 | HMGA2 | chr12:66217911-66360075 | Y | 1.00 | 3.57E-06 | 2.97 | 6.67E-08 |
| [69] | Infant head circumference | rs1042725 | |||||||
| [69] | Infant head circumference | rs7980687 | SBNO1 | chr12:123773656-123849390 | Y | −0.15 | 0.61 | −0.45 | 0.023 |
| [69] | Infant head circumference | rs11655470 | MAPT | chr17:43971748-44105700 | N | 0.61 | 0.40 | −1.82 | 0.070 |
| CRHR1 | chr17:43699267-43913194 | N | 0.15 | 0.90 | −0.13 | 0.86 | |||
| [67] | Intracranial volume | rs17689882 | CRHR1 | chr17:43699267-43913194 | N | 0.15 | 0.90 | −0.13 | 0.86 |
| [67] | Putamen volume | rs945270 | KTN1 | chr14:56025790-56168244 | Y | −0.22 | 0.67 | −0.09 | 0.80 |
| [67] | Putamen volume | rs683250 | DLG2 | chr11:83166055-85338966 | Y | 0.66 | 0.69 | −0.059 | 0.96 |
| [67] | Caudate volume | rs1318862 | FAT3 | chr11:92085262-92629618 | N | −0.92 | 2.45E-05 | −1.27 | 4.98E-06 |
| [67] | Putamen volume | rs6087771 | BCL2L1 | chr20:30252255-30311792 | Y | 0.10 | 0.89 | −0.67 | 0.029 |
| [67] | Hippocampal volume | rs61921502 | MSRB3 | chr12:65672423-65882024 | N | 0.33 | 0.79 | 2.63 | 8.69E-24 |
| [67] | Putamen volume | rs62097986 | DCC | chr18:49866542-51057784 | Y | 0.72 | 0.31 | −0.18 | 0.1 |
Significant fold changes are set in italics
DEGs are enriched with ASD and schizophrenia-risk genes
Functionally disruptive mutations in CHD8 have been reported in multiple ASD patients as well as one sporadic schizophrenia patient [72]. Comparing our list of DEGs with ASD-risk gene sets from multiple sources (see Methods), we found that upregulated genes in CHD8+/− NPCs and downregulated genes in CHD8+/− neurons were significantly enriched with ASD-risk genes (Fig. 5b, Additional file 7: Table S6). In Table 2, we provided a list of high-confident ASD-risk genes that were dysregulated in CHD8+/−. For the 163 ASD-risk genes that were in our DEG lists, they were significantly enriched for “transmission of nerve impulse,” “synaptic transmission,” and “neuron differentiation,” further supporting the importance of CHD8 in regulating synaptic functions. In a comparison of our DEGs with the genes associated with schizophrenia, we found that our DEGs were enriched with schizophrenia-related genes from the SCZgene database [58] but not enriched in the high-confident gene list from a recent schizophrenia GWAS [59] (Fig. 5b, Additional file 7: Table S6).
Table 2.
Selected differentially expressed genes associated with ASD risk
| Gene | Coordinate | ASD score | CHD8-binding in NPCs | CHD8 +/− NPC | CHD8 +/− neuron | ||||
|---|---|---|---|---|---|---|---|---|---|
| SFARI | AutismKB | Willsey | log2(FC) | q value | log2(FC) | q value | |||
| ANK2 | chr4:113739265-114304896 | High confidence | 9 | hcASD | N | −1.87 | 1.36E-45 | −1.96 | 6.32E-08 |
| SETD5 | chr3:9439299-9520924 | High confidence | 10 | Y | 0.25 | 0.67 | 0.87 | 0.00053 | |
| SUV420H1 | chr11:67922330-67981295 | High confidence | 16 | hcASD | Y | −0.084 | 0.89 | −0.86 | 0.0020 |
| SCN2A | chr2:166095912-166248818 | High confidence | 20 | hcASD | N | −0.64 | 0.86 | −3.31 | 5.67E-05 |
| DEAF1 | chr11:644233-706715 | Strong candidate | 2 | N | −0.16 | 0.79 | −0.68 | 0.0010 | |
| MYT1L | chr2:1792885-2335032 | Strong candidate | 2 | N | 0.67 | 0.72 | −1.76 | 0.0077 | |
| BCL11A | chr2:60678302-60780702 | Strong candidate | 9 | N | 1.91 | 0.029 | 0.25 | 0.61 | |
| CNTN4 | chr3:2140497-3099645 | Strong candidate | 9 | N | 6.02 | 0.53 | 5.15 | 1.60E-14 | |
| CACNA2D3 | chr3:54156574-55108584 | Strong candidate | 10 | pASD | Y | 2.64 | 0.0036 | −2.95 | 0.00076 |
| CACNA1H | chr16:1203241-1271771 | Strong candidate | 10 | Y | 0.75 | 0.045 | −0.19 | 0.58 | |
| NRXN1 | chr2:50145643-51259674 | Strong candidate | 28 | pASD | Y | −0.81 | 0.77 | −1.71 | 2.13E-05 |
| MET | chr7:116312444-116438440 | Strong candidate | 33 | N | 0.69 | 0.82 | −3.56 | 3.45E-05 | |
| GABRB3 | chr15:26788693-27184686 | Strong candidate | 34 | N | −0.43 | 0.66 | −1.70 | 0.00030 | |
| RELN | chr7:103112231-103629963 | Strong candidate | 43 | pASD | N | 1.82 | 0.16 | −2.92 | 1.46E-06 |
Significant fold changes are set in italics
CHD8-regulated genes significantly overlap with the targets of other autism-risk genes
Since ASD is caused by mutations in a diverse array of genes involved in different cellular functions, we next set out to address whether the dysregulated targets by different ASD-risk genes indeed converge on molecular and cellular pathways. We thus searched for published expression data reporting downstream targets of ASD genes, especially transcriptional regulators. Recent studies reported that in human progenitor cells, reduced expression of transcription factor 4 (TCF4) and histone-lysine N-methyltransferase 1 (EHMT1) converged at several levels with respect to their downstream targets [51]. Also, the downstream targets of methyl-CpG-binding domain 5 (MBD5) and special AT-rich binding protein (SATB2) were converged in neural stem cells [52]. We therefore decided to include these genes to our analysis because their association with ASD has been described previously [8, 73, 74], and their regulatory targets were identified in NPCs using an experimental scheme similar to ours, comparing expression changes by RNA-seq after reducing expression of ASD-candidate genes that function as transcription regulators.
First, we compared the DEGs from these studies with ours. Note that in CHD8+/− NPCs, TCF4 was upregulated (3.4-fold increase, q value = 1.58e-19), but no expression changes were observed for the other three. We found that our list of DEGs in NPCs showed significant overlap with the DEGs found in the TCF4, EHMT1, and MBD5 knockdown studies (Fig. 6a, Additional file 8: Table S7). To search for functional commonality, we analyzed the 439 genes that were affected by at least two of the five ASD genes. Again, we used STRING to define function interactions among these 439 genes. The results indicated that the common genes were mainly distributed in two highly interconnected clusters (Fig. 6b). One was significantly enriched for genes involved in forming extracellular matrix (p = 5.83e-12, Additional file 8: Table S7), including multiple collagen genes. The other was highly enriched with cell cycle-related genes (p = 6.78e-9, Additional file 8: Table S7), though the enrichment was mainly derived from the common genes between EHMT1 and MBD5 knockdown. Genes critical for neurogenesis, like PLP1 and GFAP, were also significantly enriched (p = 1.14e-8, Additional file 8: Table S7), but they showed sparse connection, perhaps because of incomplete information in the interaction database.
Fig. 6.
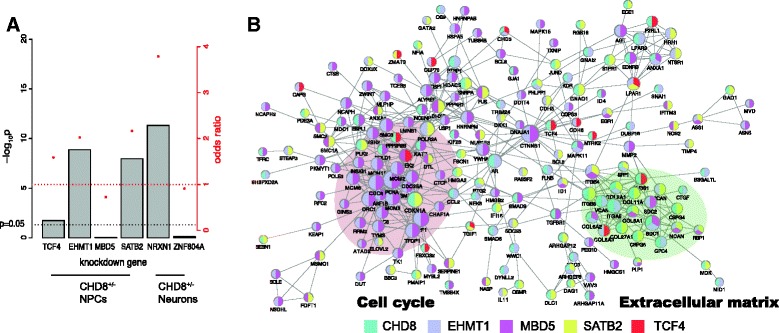
Overlap between DEGs from CHD8 +/− and previous knockdown studies of other ASD-risk genes. a Enrichment test for the overlaps between DEGs from CHD8 +/− NPCs and knockdowns of TCF4, EHMT1, MBD5, or SATB2, and between DEGs from CHD8 +/− neurons and knockdowns of ZNF804A or NRXN1. Bars represent p value (Fisher’s exact test, one-tailed), red dots represent odds ratio of overlap. b Network analysis of genes that were differentially expressed in NPCs in either CHD8 +/− or knockdown of at least two of the five genes (CHD8, TCF4, EHMT1, MBD5, and SATB2). The network was generated by STRING, with nodes representing genes and edges representing interactions. Size of the nodes is proportional to their connectivity. Colors of the nodes label the sources of DEGs. Two natural clusters are demarcated with ellipses. Disconnected genes were not shown
Next, we carried out the same analysis for DEGs in neurons, including data from our current study and a previous study in which neurexin 1 (NRXN1) expression was reduced [75]. NXRN1 was downregulated in CHD8 knockout neurons (3.2-fold reduction, q value = 2.84e-4). Again, we found a significant overlap of the DEGs from CHD8 knockout neurons with those in NRXN1 knockdown neurons (Fig. 6a). The overlapping genes were, again, enriched for “extracellular matrix.”
The transcriptional targets of zinc finger protein 804A (ZNF804A), a top schizophrenia candidate, were recently reported by our group from RNA-seq analysis in which the gene was knocked down in neurons derived from iPSCs [49]. While the overlap of DEGs from differentiating neurons with CHD8 knockout and ZNF804A knockdown was not significant (Fig. 6a), the overlap of DEGs from ZNF804A knockdown and NRXN1 knockdown was highly significant (OR = 2.63, p = 0.023, Fisher’s exact test, one-tailed). These results suggest that both common and unique transcription targets exist for neuropsychiatric disorder candidate genes that code for gene-expression regulators. It should also be noted that previous studies have also reported a convergence of genes affected by SATB2 knockdown and MBD5 knockdown [52].
In summary, our comparison of genes that show expression changes upon knockout or knockdown of key genes implicated in major neuropsychiatric disorders found that many downstream target genes were commonly affected by several transcription factors and epigenetic modifiers, and the common genes were often enriched for those that code for “cell cycle,” “extracellular matrix,” and “cell proliferation.”
Comparison with previous results from CHD8 knockdown studies
We compared our lists of DEGs with other four lists of DEGs that were obtained by knocking down CHD8 expression in neural cells [25–27]. Except for the two lists from two independent shRNA knockdowns in the same study, DEG lists from different analyses showed modest overlap; at most, 30–40 % of DEGs in one list were detected in other studies (Additional file 2: Figure S3).
Whereas the overlap from different studies was modest, we considered the possibility that the DEGs from different studies might converge on common functional pathways. We thus performed GO enrichment analysis for each of the five lists of DEGs and then focused on the 327 GO terms that were enriched in at least three DEG lists (Additional file 9: Table S8). Among them, only four terms were present in all five DEG sets (referred to as “5 L” terms) and 31 terms (“4 L” terms) were shared by four studies. When the functions of these 327 GO terms were grouped by the software GlueGO [61], however, 13 major functional clusters emerged (Fig. 7). The largest cluster, harboring 72 individual GO terms, was involved in cell communication, to which all of the 5 L terms belonged. This suggests that one common effect of reduced CHD8 expression across all studies is cell communication and signal transduction. Some of the DEGs from our CHD8+/− analysis are regulators of major signaling pathways, for example, NFATC1 and BMP2/4, and WNT7A (Fig. 3b). This finding was further supported by the observation that 62 % of the 4 L terms (n = 22) were located within similar function clusters (indicated by high density of grey lines between them), such as cellular protein metabolic process, RNA metabolic process, and cellular response to stimulus. The second largest cluster was “neuron development,” including 66 specific GO terms related to neuron differentiation, axon guidance, and synapse organization, etc. This indicates that CHD8 disruption has a profound effect on multiple aspects of neurodevelopment. Note that the two DEG lists from shRNA knockdown in the study by Cotney et al. were not enriched for this large functional cluster. However, cell cycle pathways were particularly enriched in those two lists, suggesting that the samples in that study might be in a more proliferative and less “neural-like” state. This integrated analysis also identified a number of GO terms clustered into programmed cell death and cytoskeleton organization, consistent with the previously reported interactions between CHD8 and p53-mediated apoptosis [23] and the Wnt-β-catenin signaling pathway [19]. In addition to well-known functional clusters involving CHD8, including cell adhesion and extracellular matrix organization, our bioinformatics analysis suggests that cell migration and skeletal development could also be a common theme affected by reduced CHD8 expression.
Fig. 7.
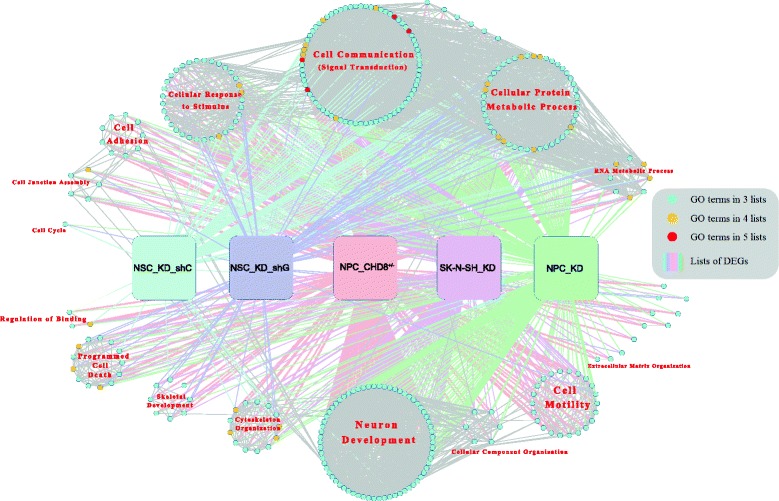
Enriched GO terms shared among five lists of DEGs from four studies. DEGs were from CHD8 +/− NPC (current data), NPC knockdown (NPC_KD) [25], NSC knockdown by two independent shRNAs (NSC_KD_shC, NSC_KD_shG) [26], or SK-N-SH knockdown (SK-N-SH_KD) [27]. Nodes represent common GO terms (names not displayed) and grey edges between them depict the degrees of gene overlap between terms. GO Terms enriched in 3, 4, and 5 DEG lists are colored blue, orange, and red, respectively. Individual GO terms with a similar gene composition were clustered into functional groups by ClueGO and drawn together as circles, with the functions labeled in red font. Square boxes represent each of the five DEG lists and colored edges connect enriched GO terms in individual studies. This visualization was created using ClueGO plugin in the Cytoscape
Finally, we compared the five lists of DEGs to the results in a recent analysis of telencephalic organoids generated from idiopathic autism patient-specific iPSCs [76]. Of the differentially expressed genes in two developmental stages (TD11 and TD31) of the organoids between ASD and control samples [76], 516 and 1060 were expressed in our samples. When the DEGs in CHD8+/− or CHD8 knockdown studies were compared to the organoid DEGs, our CHD8+/− showed the largest overlap with the organoid data (Fig. 8, Additional file 2: Figure S3). GO analysis indicated that the dysregulated genes common in CHD8+/− samples and ASD organoids were significantly enriched for genes involved in skeleton system development, cell adhesion, and neuron differentiation (Fig. 8), indicating that these could be functional pathways commonly disrupted in ASD. Interestingly, the brain volume-associated gene FAT3 was upregulated in TD31, further suggesting the potential importance of FAT3 in ASD.
Fig. 8.
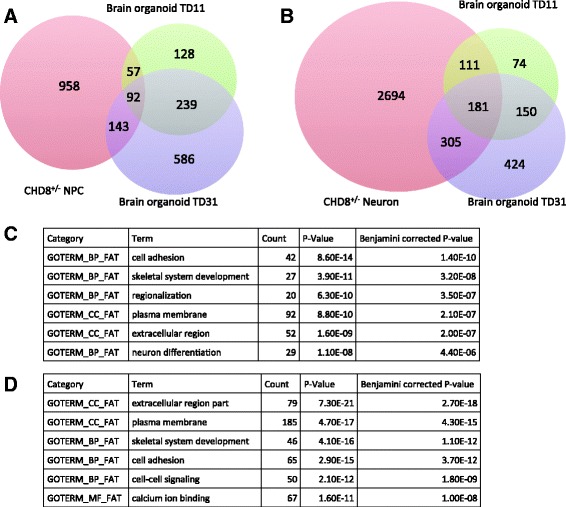
Comparison of DEGs from CHD8 +/− and DEGs from brain ASD organoids [76]. Venn diagrams of DEGs from CHD8 +/− NPCs (a) or neurons (b) and DEGs from brain organoids. The number in each section represents genes that are also expressed in our samples. Top-enriched GO terms for overlapped genes between DEGs from CHD8 +/− NPCs (c) or neurons (d) and DEGs from brain organoids referred to as TD11 and TD31
Discussion
To better understand the effect of genetic disruption of CHD8 in ASD subjects, we have applied CRISPR/Cas9 technology to knockout one copy of CHD8 in a control iPSC line and studied its effect on gene expression during early neurodevelopment. In comparison to previous studies [25–27] that used a gene knockdown approach to reduce CHD8 expression, our approach generated heterozygous disruptions that better mimic the germline mutations in ASD patients and allows for the study of long-term effects of CHD8 disruption in neurogenesis in vitro. In addition, creating CHD8+/− iPSCs provides a truly renewable resource for investigators, as opposed to NPCs, which have a finite replicative capacity. Furthermore, iPSCs can differentiate into any cell type, including cerebral organoids [77]; NPCs are restricted in their differentiation potential.
Perhaps one of the most important findings emerging from our transcriptomic analysis is that several genes known to be related to head size or brain volume, either from the analysis of human phenotype ontology [46] or identified through GWAS [67–69], displayed significant changes in their expression in CHD8+/− neurons. Studies examining CHD8 function in zebrafish during embryonic development revealed that the macrocephaly phenotype observed in ASD probands with CHD8 mutations is likely caused by disturbed neuronal proliferation at early developmental stages [15, 25]. By uncovering genes like HMGA2 and FAT3, which have been associated with head size, our finding thus provides new molecular insights that may eventually link CHD8 mutation to abnormal neuronal proliferation and macrocephaly. This association between macrocephaly and ASD is not the first to be reported in genetically defined subgroups. Mutations in PTEN are also associated with severe macrocephaly and ASD [78]. Although PTEN itself was not differentially expressed in our CHD8+/− samples, FOXO1 and FOXO3, two critical transcription factors in PTEN signaling, were. Interestingly, IPA analysis of the differentially expressed genes reported an enrichment of genes in PTEN signaling, suggesting that there may be a link between the PTEN and CHD8 pathways, and the molecular link underlies the observed macrocephaly and ASD. In this regard, we should mention that PTEN was differentially expressed upon CHD8 knockdown in NPCs in a previous study [25]. We should also point out there were additional genes in our DEG lists that have been previously suggested to be associated with hippocampal volumes, such as BDNF, DISC1, and NRG1.
Dysregulated genes in CHD8+/− cells exhibited significant overlap with previously defined ASD-risk genes, including some high confident candidates like ANK2, SCN2A, and SUV420H1 [54] that also showed significant differential expression (Table 2). We further demonstrated, interestingly, that CHD8-regulated genes significantly overlapped with the downstream targets of other ASD-risk genes like TCF4 and NRXN1, providing transcriptomic evidence that ASD-risk genes have overlapping function and converge on downstream regulatory pathways. This is extremely important in a genetically heterogeneous condition, such as ASD, in which any individual candidate gene carrying deleterious mutation may only contribute to 1~2 % ASD cases [79]. TCF4 is associated with Pitt-Hopkins syndrome (MIM: 610954), which is defined by severe psychomotor delay, epilepsy, daily bouts of diurnal hyperventilation, mild postnatal growth retardation, postnatal microcephaly, and distinctive facial features [80]. It is also a top schizophrenia candidate gene. In NPCs and NSCs, CHD8 binds to the TCF4 gene body in a region that is also enriched with H3K27ac [25, 26], a histone modification associated with active enhancers. TCF4 is significantly upregulated in both CHD8+/− NPCs and neurons. Moreover, TCF4-regulated genes significantly overlapped with CHD8-regulated genes. Taken together, these results suggest a direct connection between TCF4 and CHD8 regulatory networks. The common genes in the two networks, especially those regulated oppositely by CHD8 and TCF4, such as HMGA2, which was upregulated in CHD8+/− (Table 1) and downregulated in TCF4 knockdown [81], are strong candidates for regulating the development of brain size.
It was intriguing to find that DEG lists from different CHD8+/− or knockdown studies had only limited overlaps. A recent study of knockout vs. knockdown in zebrafish egfl7 proposed that compensatory networks would be activated to buffer against deleterious mutations from knockout, which was absent in knockdown [82], providing a potential explanation for the lack of good overlap among CHD8 KO and KD findings. However, we found that at the function and pathway levels, genes involved in similar functions were affected by reduced CHD8 expression in different contexts. It is conceivable that a limited number of upstream regulators are directly regulated by CHD8, and the subsequent response of the downstream targets is mostly dependent on genetic background, cell culture conditions, and other experimental factors. In this regard, we should mention that five (GDPD4, VPS13B, KMT2C, SETBP1, and CLTCL1) of the ~1000 ASD-risk genes were predicted to be functionally disrupted by premature stop or frameshift variants located to the coding exons of the subject used to prepare our WT iPSC line (Additional file 10: Table S9 and S10). However, none of these five genes exhibited differential expression in the WT neurons when compared to samples from other control iPSC lines derived from six unrelated subjects (data not shown). As neuronal differentiation is a complex process, affected by both environmental cues and intrinsic cellular signaling, our analysis suggests that it is important to study the effects of the same genes under different experimental conditions. Nevertheless, comparison of our results with the transcriptomic data in ASD-derived organoids indicates that reduction of CHD8 expression in our CHD8+/− samples is probably more consistent with the gene regulation in ASD-developing brains, although it should be noted that the ASD-derived organoids were from patients with unknown genetic mutations.
As most of the functionally disruptive mutations uncovered in the CHD8-coding regions in the ASD probands introduce premature stop or frameshift mutation [15, 16], we used CRISPR/Cas9 technology to make small deletions in the first coding exon of CHD8 in this study. While the 2- or 10-bp deletion is predicted to cause frameshift, and no functional protein is expected from the mutants, RNA transcripts from the knockout CHD8 copy were observed in our RNA-seq data, indicating nonsense mediated mRNA decay is incomplete if it occurs to the mutated CHD8 transcripts (Fig. 1). CHD8 encodes a multi-domain protein, and deleterious mutations in CHD8 have been found in almost every important functional domain [15, 16]. While our data indicate that CHD8 regulates multiple pathways related to neural development, the different mutations in ASD individuals may impair distinct and specific aspects of CHD8 functions. In the future, it will be valuable to carry out gene-expression profiling using ASD-specific iPSCs to see how our current findings can be recapitulated in additional iPSC-derived NPCs and neurons. As genetic background likely plays an important role in modulating CHD8 function during brain development, it is important to both perform our current knockout analysis in additional control iPSC lines and to derive patient-specific lines from multiple ASD individuals with CHD8 mutations. In addition, it will be extremely valuable to apply CRISPR technology to correct the CHD8 mutations and perform transcriptomic analysis and other molecular assays once the patient-specific lines are established.
Conclusions
CHD8 regulates multiple genes involved in cell communication, extracellular matrix and neurogenesis that are critical for brain development. In addition, CHD8 hemizygosity causes expression changes in several genes that are associated with brain volume or head size, suggesting a molecular link between CHD8 mutation and macrocephaly. By cross-analysis of several studies in which the expression of several ASD-candidate genes was reduced, we provide evidence that the transcription targets of ASD genes converge on a set of genes and pathways.
Acknowledgements
We would like to thank Dr. Brett Abrahams for his critical comments on the manuscript. We also would like to thank Dr. Carl Ernst and Dr. Derek Blake for sharing DEG lists of TCF4, EHMT1, MBD5, and SATB2 knockdown and the Einstein High Performance Computing for computing support. This work was supported by grants from the National Institute of Mental Health (NIMH; MH099452 to DZ, and MH099427 and MH087840 to HML). WG would like to acknowledge the support from NYSTEM (C028109 and C029571). This work was also supported in part by a grant to The Rose F. Kennedy Intellectual and Developmental Disabilities Research Center (RFK-IDDRC) from the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD) at the NIH (1P30HD071593-01).
Abbreviations
- CHD8
Chromodomain helicase DNA binding protein 8
- CNV
Copy number variation
- DEG
Differentially expressed gene
- FPKM
Fragments per kilobase of exon per million fragments mapped
- GO
Gene ontology
- GWAS
Genome-wide association study
- IPA
Ingenuity pathway analysis
- iPSC
Induced pluripotent stem cell
- KD
Knockdown
- KO
Knockout
- NPC
Neural progenitor cell
- NSC
Neural stem cell
- OR
Odds ratio
- sgRNA
Single guide RNA
- WT
Wild-type
Additional files
Supplemental methods. (PDF 101 kb)
Figure S1–S3 and Table S1. (PDF 367 kb)
Table S2. Full list of gene-expression values, fold changes, and statistics. (XLSX 562 kb)
Table S3. GO analysis (DAVID) of DEGs in NPCs and neurons (only terms with Benjamini corrected p < 0.05 were included). (XLSX 155 kb)
Table S4. Pathway analysis (IPA) of DEGs in NPCs and neurons (only pathways with Benjamini corrected p < 0.05 were included). (XLSX 177 kb)
Table S5. Human phenotype analysis (Toppgene) of DEGs. (XLSX 153 KB)
Table S6. DEGs associated with ASD or schizophrenia. (XLSX 49 kb)
Table S7. DEGs from CHD8 +/− or knockdown of TCF4, EHMT1, MBD5, SATB2, NRXN1, or ZNF804A expression (“1” represents differential expression, “0” represents no change) and GO analysis from the STRING (only term with FDR < 0.05 were included). (XLSX 292 kb)
Table S8. GO analysis (ClueGO) of DEGs in each of five DEG lists from CHD8 +/− or knockdown analysis. (XLSX 670 kb)
Table S9–S10. Numbers and putative functionally disrupted coding variants in the WT iPSC line. (PDF 34 kb)
Footnotes
Ping Wang and Mingyan Lin shared first authorship.
Competing interests
The authors declare no competing financial interests and no non-financial conflicts of interest for any of the authors.
Authors’ contributions
HML and DZ conceived of the project. HML performed experimental data analysis. PW, ML, and DZ performed the bioinformatics data analysis. EP and AH prepared the iPS lines, neuronal differentiation, RNA-seq samples, and qPCR validation. WG designed the CRISPR guide sequences. ZZ and WG performed the Western blot analysis. PW, ML, HML, and DZ wrote the manuscript. All authors edited and approved the final manuscript.
Contributor Information
Ping Wang, Email: ping.wang@einstein.yu.edu.
Mingyan Lin, Email: mingyan.lin@phd.einstein.yu.edu.
Erika Pedrosa, Email: erika.pedrosa@einstein.yu.edu.
Anastasia Hrabovsky, Email: anastasia.hrabovsky@gmail.com.
Zheng Zhang, Email: zheng.zhang@einstein.yu.edu.
Wenjun Guo, Email: wenjun.guo@einstein.yu.edu.
Herbert M. Lachman, Email: herb.lachman@einstein.yu.edu
Deyou Zheng, Email: deyou.zheng@einstein.yu.edu.
References
- 1.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5) (American Psychiatric Association, 2013).
- 2.De Rubeis S, Buxbaum JD. Genetics and genomics of autism spectrum disorder: embracing complexity. Hum Mol Genet. 2015;24:R24–31. doi: 10.1093/hmg/ddv273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151:1431–1442. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2013;93:249–263. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015;47:582–588. doi: 10.1038/ng.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krumm N, O'Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014;37:95–105. doi: 10.1016/j.tins.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158:263–276. doi: 10.1016/j.cell.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaste P, Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, et al. Adjusting head circumference for covariates in autism: clinical correlates of a highly heritable continuous trait. Biol Psychiatry. 2013;74:576–584. doi: 10.1016/j.biopsych.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marfella CG, Imbalzano AN. The Chd family of chromatin remodelers. Mutat Res. 2007;618:30–40. doi: 10.1016/j.mrfmmm.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishiyama M, Skoultchi AI, Nakayama KI. Histone H1 recruitment by CHD8 is essential for suppression of the Wnt-beta-catenin signaling pathway. Mol Cell Biol. 2012;32:501–512. doi: 10.1128/MCB.06409-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson BA, Tremblay V, Lin G, Bochar DA. CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol Cell Biol. 2008;28:3894–3904. doi: 10.1128/MCB.00322-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawada G, Ueo H, Matsumura T, Uchi R, Ishibashi M, Mima K, et al. CHD8 is an independent prognostic indicator that regulates Wnt/beta-catenin signaling and the cell cycle in gastric cancer. Oncol Rep. 2013;30:1137–1142. doi: 10.3892/or.2013.2597. [DOI] [PubMed] [Google Scholar]
- 22.Okerlund ND, Cheyette BN. Synaptic Wnt signaling-a contributor to major psychiatric disorders? J Neurodev Disord. 2011;3:162–174. doi: 10.1007/s11689-011-9083-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishiyama M, Oshikawa K, Tsukada Y, Nakagawa T, Iemura S, Natsume T, et al. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat Cell Biol. 2009;11:172–182. doi: 10.1038/ncb1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subtil-Rodriguez A, Vazquez-Chavez E, Ceballos-Chavez M, Rodriguez-Paredes M, Martin-Subero JI, Esteller M, et al. The chromatin remodeller CHD8 is required for E2F-dependent transcription activation of S-phase genes. Nucleic Acids Res. 2014;42:2185–2196. doi: 10.1093/nar/gkt1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P, et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci U S A. 2014;111:E4468–4477. doi: 10.1073/pnas.1405266111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun. 2015;6:6404. doi: 10.1038/ncomms7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkinson B, Grepo N, Thompson BL, Kim J, Wang K, Evgrafov OV, et al. The autism-associated gene chromodomain helicase DNA-binding protein 8 (CHD8) regulates noncoding RNAs and autism-related genes. Transl Psychiatry. 2015;5:e568. doi: 10.1038/tp.2015.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ceballos-Chavez M, Subtil-Rodriguez A, Giannopoulou EG, Soronellas D, Vazquez-Chavez E, Vicent GP, et al. The chromatin Remodeler CHD8 is required for activation of progesterone receptor-dependent enhancers. PLoS Genet. 2015;11:e1005174. doi: 10.1371/journal.pgen.1005174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez-Paredes M, Ceballos-Chavez M, Esteller M, Garcia-Dominguez M, Reyes JC. The chromatin remodeling factor CHD8 interacts with elongating RNA polymerase II and controls expression of the cyclin E2 gene. Nucleic Acids Res. 2009;37:2449–2460. doi: 10.1093/nar/gkp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao D, Lin M, Chen J, Pedrosa E, Hrabovsky A, Fourcade HM, et al. MicroRNA Profiling of Neurons Generated Using Induced Pluripotent Stem Cells Derived from Patients with Schizophrenia and Schizoaffective Disorder, and 22q11.2 Del. PLoS One. 2015;10:e0132387. doi: 10.1371/journal.pone.0132387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin M, Hrabovsky A, Pedrosa E, Wang T, Zheng D, Lachman HM. Allele-biased expression in differentiating human neurons: implications for neuropsychiatric disorders. PLoS One. 2012;7:e44017. doi: 10.1371/journal.pone.0044017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin M, Pedrosa E, Shah A, Hrabovsky A, Maqbool S, Zheng D, et al. RNA-Seq of human neurons derived from iPS cells reveals candidate long non-coding RNAs involved in neurogenesis and neuropsychiatric disorders. PLoS One. 2011;6:e23356. doi: 10.1371/journal.pone.0023356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.CRISPR design [http://crispr.mit.edu/]
- 37.Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Picard [http://broadinstitute.github.io/picard/]
- 41.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 44.da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.IPA [http://www.qiagen.com/ingenuity]
- 46.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2079;25:2078. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J, Lin M, Foxe JJ, Pedrosa E, Hrabovsky A, Carroll R, et al. Transcriptome comparison of human neurons generated using induced pluripotent stem cells derived from dental pulp and skin fibroblasts. PLoS One. 2013;8:e75682. doi: 10.1371/journal.pone.0075682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen J, Lin M, Hrabovsky A, Pedrosa E, Dean J, Jain S, et al. ZNF804A Transcriptional Networks in Differentiating Neurons Derived from Induced Pluripotent Stem Cells of Human Origin. PLoS One. 2015;10:e0124597. doi: 10.1371/journal.pone.0124597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen ES, Gigek CO, Rosenfeld JA, Diallo AB, Maussion G, Chen GG, et al. Molecular convergence of neurodevelopmental disorders. Am J Hum Genet. 2014;95:490–508. doi: 10.1016/j.ajhg.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gigek CO, Chen ES, Ota VK, Maussion G, Peng H, Vaillancourt K, et al. A molecular model for neurodevelopmental disorders. Transl Psychiatry. 2015;5:e565. doi: 10.1038/tp.2015.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.SFARI [https://gene.sfari.org/autdb/GS_Home.do]
- 55.Xu LM, Li JR, Huang Y, Zhao M, Tang X, Wei L. AutismKB: an evidence-based knowledgebase of autism genetics. Nucleic Acids Res. 2012;40:D1016–1022. doi: 10.1093/nar/gkr1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu L, Lei J, Sanders SJ, Willsey AJ, Kou Y, Cicek AE, et al. DAWN: a framework to identify autism genes and subnetworks using gene expression and genetics. Mol Autism. 2014;5:22. doi: 10.1186/2040-2392-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- 59.Schizophrenia Working Group of the Psychiatric Genomics C Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 61.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.R [http://www.R-project.org/]
- 64.Novara F, Beri S, Giorda R, Ortibus E, Nageshappa S, Darra F, et al. Refining the phenotype associated with MEF2C haploinsufficiency. Clin Genet. 2010;78:471–477. doi: 10.1111/j.1399-0004.2010.01413.x. [DOI] [PubMed] [Google Scholar]
- 65.Chakrabarti B, Dudbridge F, Kent L, Wheelwright S, Hill-Cawthorne G, Allison C, et al. Genes related to sex steroids, neural growth, and social-emotional behavior are associated with autistic traits, empathy, and Asperger syndrome. Autism Res. 2009;2:157–177. doi: 10.1002/aur.80. [DOI] [PubMed] [Google Scholar]
- 66.King IF, Yandava CN, Mabb AM, Hsiao JS, Huang HS, Pearson BL, et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501:58–62. doi: 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivieres S, Jahanshad N, et al. Common genetic variants influence human subcortical brain structures. Nature. 2015;520:224–229. doi: 10.1038/nature14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stein JL, Medland SE, Vasquez AA, Hibar DP, Senstad RE, Winkler AM, et al. Identification of common variants associated with human hippocampal and intracranial volumes. Nat Genet. 2012;44:552–561. doi: 10.1038/ng.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taal HR, St Pourcain B, Thiering E, Das S, Mook-Kanamori DO, Warrington NM, et al. Common variants at 12q15 and 12q24 are associated with infant head circumference. Nat Genet. 2012;44:532–538. doi: 10.1038/ng.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deans MR, Krol A, Abraira VE, Copley CO, Tucker AF, Goodrich LV. Control of neuronal morphology by the atypical cadherin Fat3. Neuron. 2011;71:820–832. doi: 10.1016/j.neuron.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry. 2014;19:652–658. doi: 10.1038/mp.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van Balkom ID, Vuijk PJ, Franssens M, Hoek HW, Hennekam RC. Development, cognition, and behaviour in Pitt-Hopkins syndrome. Dev Med Child Neurol. 2012;54:925–931. doi: 10.1111/j.1469-8749.2012.04339.x. [DOI] [PubMed] [Google Scholar]
- 74.Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeng L, Zhang P, Shi L, Yamamoto V, Lu W, Wang K. Functional impacts of NRXN1 knockdown on neurodevelopment in stem cell models. PLoS One. 2013;8:e59685. doi: 10.1371/journal.pone.0059685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell. 2015;162:375–390. doi: 10.1016/j.cell.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin M, Zhao D, Hrabovsky A, Pedrosa E, Zheng D, Lachman HM. Heat shock alters the expression of schizophrenia and autism candidate genes in an induced pluripotent stem cell model of the human telencephalon. PLoS One. 2014;9:e94968. doi: 10.1371/journal.pone.0094968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abrahams BS, Geschwind DH. Connecting genes to brain in the autism spectrum disorders. Arch Neurol. 2010;67:395–399. doi: 10.1001/archneurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Amiel J, Rio M, de Pontual L, Redon R, Malan V, Boddaert N, et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet. 2007;80:988–993. doi: 10.1086/515582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Forrest MP, Waite AJ, Martin-Rendon E, Blake DJ. Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation. PLoS One. 2013;8:e73169. doi: 10.1371/journal.pone.0073169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rossi A, Kontarakis Z, Gerri C, Nolte H, Holper S, Kruger M, et al. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524:230–233. doi: 10.1038/nature14580. [DOI] [PubMed] [Google Scholar]