

A cell in a complex organism must balance differentiation, proliferation, and specialized functions with readiness to thwart pathogens and uncontrolled growth. The intricacy of balancing cell fates is exemplified by the interplay of signals from Ras versus interferon pathways.
The small Ras GTPases are eminent oncogenes and master regulators of countless cellular processes that promote cell proliferation, differentiation and survival. Interferon-β (IFN-β) is a cytokine produced during virus infection or cell damage and a potent stimulant of antiviral and antitumor responses. Ras- vs. IFN- signaling establish opposing fates for a cell: growth promotion versus cessation. Intriguingly, overactive H-Ras signaling can reduce IFN-β expression and response to IFN-β which is exploited by some oncolytic viruses that specifically replicate in tumor over healthy cells. For example, reovirus is a normally harmless virus that inherently replicates in IFN signaling-deficient tumor cells and offers a candidate oncotherapy.
It is unclear why H-Ras and IFN pathways interlink, but perhaps Ras signaling inadvertently activates IFNs, which must then be subdued. A more tangible question is how H-Ras pathways muzzle IFN-β. Cells are equipped with proteins that detect foreign pathogen- or damage- associated molecular patterns (PAMPs and DAMPs). These sensors activate a cascade of signaling molecules that lead to phosphorylation and nuclear translocation of IFN-regulatory factors (IRFs). Expression of IFN-β is regulated by IRFs, along with other transcription factors (ATF2/c-jun and NFκB) shared with pathways such as Ras. Secreted IFN-β leads to autocrine and paracrine activation of STAT and IRF transcription factors that drive expression of hundreds of IFN-stimulated genes (ISGs) and establish a state non-conducive to virus replication.
Previous studies implicated the MEK/ERK pathway downstream of Ras in suppressing expression of IFNs and ISGs, thereby promoting susceptibility to oncolytic reovirus.7 H-Ras transformation was associated with increased levels of STAT1, STAT2, and IRF9.3 Komatsu et al4 discovered that MEK/ERK signaling downregulates IRF1 and subsequent expression of ISGs, and that introduction of IRF1 re-established IFN expression in cancer cells. These findings support a link between Ras/MEK/ERK and transcription factors necessary for IFN-β and ISG expression.
Ahn et al.1 introduce a new prominent but unexpected player to the Ras-IFN-reovirus connection (Fig. 1). As an unbiased approach to discover changes associated with Ras transformation, the authors biotinylated cell-surface proteins of immortalized mouse fibroblasts in the presence or absence of constitutively active H-RasG12V. Nogo-B was a striking contender; H-Ras transformation corresponded with a dramatic loss of full length Nogo-B and accumulation of an N-terminally cleaved Nogo-B product.
RNAi-mediated silencing of the Nogo gene significantly increased reovirus-induced IFN-β and ISG expression in non- and especially Ras-transformed cells. Conversely, over-expression of Nogo-B reduced ISG expression. Similar trends were observed when poly(I:C) was used as an IFN stimulant. These findings suggest a causal relationship between Nogo-B and inhibition of IFN signaling. Truncated Nogo-B appears to exert higher IFN-inhibitory function; a possibility that should be confirmed by comparing activities of exogenously-introduced full-length vs. truncated Nogo-B.
Previous studies found that Nogo-B is subject to phosphorylation by ERKs and cyclin-dependent kinases (cdks), and can undergo caspase-7 mediated cleavage.6 In the context of restricting IFN and ISG expression, Ahn et al found a distinct Nogo-B cleavage product that is generated independently of MEK and caspase. The contribution of cdks and Nogo-B phosphorylation remains to be tested.
Though aptly named for its new role, Nogo-B is not an intuitive player in IFN signaling. Nogo-A, Nogo-B, and Nogo-C are 3 integral membrane protein isoforms generated from the reticulon 4 gene. The best studied isoform Nogo-A is involved in neurite outgrowth inhibition. Roles for Nogo proteins outside of the CNS are poorly understood.5
The Nogo-B isoform resides at the plasma membrane and intracellularly in both neuronal and non-neuronal cells.2 Is Nogo-B affecting IFN directly, for example by modulating assembly of IFN signaling molecules? Or does it function indirectly, driving other pathways that converge on IFN? How does removal of the acidic and proline-rich N-terminus of Nogo-B promote IFN inhibition, and does it relate to altered association with Nogo-B receptors? How do signaling pathways downstream of H-Ras promote Nogo-B cleavage, do other isoforms of Ras have similar effects, and what extent of Ras activity is sufficient? Are there additional effects of Nogo-B cleavage on transformation? Do Nogo-B levels reflect efficiency of virus replication in vitro and in vivo, and in what cell contexts? The current study supports further consideration of Nogo-B in shaping cell fates outside of the nervous system.
Figure 1.
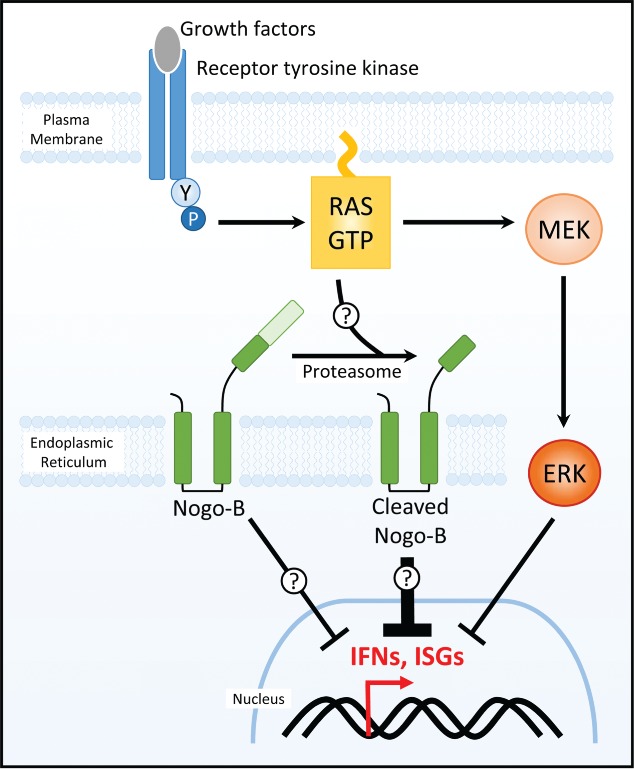
Ahn et al. identify Nogo-B as a novel modulator of interferon signaling. Activated Ras caused pronounced proteasome-dependent Nogo-B cleavage (ER-bound Nogo-B is depicted in this schematic that is likely also applicable to plasma membrane-bound Nogo-B). Both full-length and cleaved Nogo-B contributed to reduced interferon signaling, with cleaved Nogo-B having more potent inhibitory effects. Nogo-B regulated interferon suppression occurred independent of the well-implicated Ras/MEK/ERK pathway. Detailed mechanistic links between Ras, Nogo-B, and IFN remain to be established.
References
- 1.Ahn DG, et al.. Cell Cycle 2015; 14(14):2301-10; PMID:25946643; http://dx.doi.org/ 10.1080/15384101.2015.1044187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dodd DA, et al. J Biol Chem 2005; 280(13):12494; PMID:15640160; http://dx.doi.org/ 10.1074/jbc.M411827200 [DOI] [PubMed] [Google Scholar]
- 3.Klampfer L, et al. J Biol Chem 2003; 278(47):46278-87; PMID:12972432; http://dx.doi.org/ 10.1074/jbc.M304721200 [DOI] [PubMed] [Google Scholar]
- 4.Komatsu Y, et al.. Oncogene 2014; PMID:25347735; http://dx.doi.org/21045861 10.1038/onc.2014.331 [DOI] [Google Scholar]
- 5.Schwab ME. Nat Rev Neurosci 2010; 11(12):799-811; PMID:21045861; http://dx.doi.org/ 10.1038/nrn2936 [DOI] [PubMed] [Google Scholar]
- 6.Schweigreiter R, et al. Proteomics 2007; 7(24):4457-67; PMID:18072206; http://dx.doi.org/ 10.1002/pmic.200700499 [DOI] [PubMed] [Google Scholar]
- 7.Shmulevitz M, et al. Cancer Res 2010; 70(12):4912-21; PMID:20501842; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-4676 [DOI] [PubMed] [Google Scholar]