

Abstract
HIV-1 Vif assembles the Cul5-EloB/C E3 ubiquitin ligase to induce proteasomal degradation of the cellular antiviral APOBEC3 proteins. Detailed structural studies have confirmed critical functional domains in Vif that we have previously identified as important for the interaction of EloB/C, Cul5, and CBFβ. However, the mechanism by which Vif recognizes substrates remains poorly understood. Specific regions of Vif have been identified as being responsible for binding and depleting APOBEC3G and APOBEC3F. Interestingly, we have now identified distinct yet overlapping domains that are required for HIV-1 Vif-mediated G2/M-phase cell cycle arrest and APOBEC3H degradation, but not for the inactivation of APOBEC3G or APOBEC3F. Surprisingly, Vif molecules from primary HIV-1 variants that caused G2/M arrest were unable to inactivate APOBEC3H; on the other hand, HIV-1 Vif variants that could inactivate APOBEC3H were unable to induce G2/M arrest. All of these Vif variants still maintained the ability to inactivate APOBEC3G/F. Thus, primary HIV-1 variants have evolved to possess distinct functional activities that allow them to suppress APOBEC3H or cause G2 cell cycle arrest, using mutually exclusive interface domains. APOBEC3H depletion and G2 arrest are apparently evolutionary selected features that cannot co-exist on a single Vif molecule. The existence and persistence of both types of HIV-1 Vif variant suggests the importance of APOBEC3H suppression and cell cycle regulation for HIV-1's survival in vivo.
Keywords: APOBEC3H, cell cycle regulation, evolutionary selection, proteasomal degradation, Vif
Introduction
Viral infectivity factor (Vif) is required for human immunodeficiency virus type 1 (HIV-1) replication in primary cells.1,2 Vif overcomes host restriction factors, such as APOBEC3G, to promote viral infection.3-5 It specifically induces proteasomal degradation of APBOEC3 proteins, including APOBEC3G5-7 and APOBEC3F.8-11 For this purpose, Vif hijacks host cellular factors, including Cul5, EloB, EloC, and Rbx, and forms a Cullin ring-based ubiquitin ligase (also known as the E3 complex).5,12,13 Once the E3 complex is assembled, Vif interacts with the APOBEC3 proteins and induces their ubiquitination and degradation;5,7,9,11 thereby, Vif promotes the production of infectious HIV-1 progeny in certain types of cells.14 We more recently determined that another cellular factor, CBFβ, is also critical for the formation of the Vif-induced E3 complex. Without CBFβ, Vif interacts only with EloC and EloB, but not with Cul5; thus, it fails to assemble a functional E3 complex and leaves APOBEC3 proteins intact.15,16 In addition to its importance for molecular virology research, CBFβ also stabilizes the Vif protein in prokaryotic E.coli cells.17,18
The HIV-1 NL4–3 Vif (VifNL4–3) structure recently revealed by Guo et al. verifies several previous observations about Vif-induced E3 complex formation.19 CBFβ, as the essential component promoting the assembly of the Vif-E3 complex,15 actually becomes a subunit of the E3 complex. The potent Vif-CBFβ interaction involves multiple regions on both proteins, including CBFβ 68–73 directly interacting with Vif;20,21 CBFβ 121–126, in a β-sheet, possibly maintaining the structure of CBFβ;20 the Vif N-terminus inserting itself into the CBFβ protein;18 and several conserved residues being important for Vif-CBFβ binding.18,22 Also, Vif interacts with EloB/C through the B/C box, whose previously determined 144SLQ146 motif5,12,13 contributes most significantly to the interaction. Surprisingly, although several regions of Vif have been identified to be critical for Cul5 binding,23-29 only one α-helix is utilized for direct interaction with Cul5, but the importance of the Vif HCCH domain9,23,24,30,31 and zinc binding25-27 have been verified by the structure of the complex. The essential requirement of EloB/C for the Vif-induced E3 complex assembly5 has also been demonstrated by the binding of Vif and Cul5 to the complex, which facilitates their interaction with each other.
Although the structural details of the Vif-E3 complex have now been revealed, the Vif-substrate interaction is still not fully understood. Interestingly, Vif interacts with APOBEC3 proteins through the N-terminus of Vif, but different regions/motifs apply for each interaction, based on the specific substrate. Collectively, Vif positions Vif 23SLV/Ix4Yx9Y40 and 52VxIPLx4–5LxΦx2YWxL72 (where "x" stands for any amino acid residue, "Φ" for a hydrophobic residue) have been proved important for interactions with both APOBEC3F and APOBEC3G as well as their depletion;32-34 position 11 and the motifs 14DRMR17 and 74TGERxW79 are critical for APOBEC3F binding, and 40YRHHY44 for APOBEC3G binding.32,34-36 Consistently, the revealed structure of Vif has confirmed that the motifs for both APOBEC3F and APOBEC3G (above) are critical for maintaining Vif structure, while the latter residues specific for either APOBEC3F or APOBEC3G degradation are located in 2 different surface areas on Vif,19 thus are more possibly involved in direct APOBEC3F or APOBEC3G interaction.
As compared to the comprehensive studies that have been conducted regarding the Vif-APOBEC3F/G interaction, only a little information has been reported concerning Vif-APOBEC3H binding. There are several APOBEC3H haplotypes in human cells, and some of them, such as hap II, are stably expressed and are potent inhibitors of HIV-1 replication.37-39 Interestingly, in contrast to the universal effect against APOBEC3F/G, only some Vifs can induce the depletion of APOBEC3H, most likely because of the presence of antiviral APOBEC3H haplotype(s) that causes selection pressure.40 We recently discovered 2 different positions, 39 and 48, on HIV-1 Vif in which mutations greatly affect the capacity to degrade APOBEC3H without compromising APOBEC3F/G depletion,40-42 indicating that different regions of the Vif protein are critical for APOBEC3F/G/H interactions and/or depletion.
In addition to depleting the APOBEC3 protein, Vif also functions as a cell cycle regulator, arresting host cells at the G2/M phage (G2 arrest).43,44 This process also requires the presence of CBFβ and the Vif-induced E3 complex (unpublished data, manuscript submitted). It is worth noting that, as for APOBEC3H depletion, Vif-induced G2 arrest is applicable only to some Vif proteins.45 Indeed, a comparison between HXB2 and NL4–3 Vif has led to the discovery of positions 31, 33, 36, 47, and 50 as being critical for Vif-induced G2 arrest, whereas alteration of all 5 residues gives VifHXB2 potency in G2 arrest.45 The fact that APOBEC3H depletion and G2 arrest are the only 2 known functions that appear to be selective among Vif proteins suggests that there may be a relationship between these 2 functions.
In the present study, we screened several Vif proteins from different HIV-1 strains for their potency in APOBEC3H removal and G2 arrest. When combined with our previously published data, our findings reveal that no single Vif protein can exhibit both functions. Vif structure modeling indicated that the residues that are reported to be critical for APOBEC3H removal or G2 arrest are located in 2 similar regions of the Vif protein, indicating that there may be competition for binding to one factor or the other, leading to Vif's variants showing a preference for one of the functions over the other. Indeed, exchanging the critical residues in Vif proteins can alter their potency in removing APOBEC3H and/or regulating the cell cycle. With the detection of a new G2 arrest-competent VifC2, we have excluded Vif position 47 from the list of residues that are critical for Vif-induced cell cycle regulation, but have revealed this residue's importance for Vif-mediated APOBEC3H depletion. Therefore, our data demonstrate that APOBEC3H depletion and cell cycle regulation are 2 selective features that cannot co-exist in a single Vif protein, and they have revealed additional essential residues for both functions, which will certainly provide further insight into Vif's involvement in the pathogenesis and general virology of HIV-1.
Results
HIV-1 Vif proteins that are competent for APOBEC3H depletion are defective for inducing G2/M cell cycle arrest
It has previously been shown that VifNL4–3 causes cell cycle delay in multiple cell lines.43-45 We also experimentally confirmed that VifNL4–3 could induce G2 arrest in human embryo kidney 293T (HEK293T) cells (Fig. 1A, B). In contrast, VifHXB2 failed to cause G2 arrest in HEK293T cells (Fig. 1A, B). Interestingly, VifHXB2 and VifNL4–3 showed a converse potency in APOBEC3H depletion and G2 arrest in HEK293T cells: Indeed, VifHXB2, which was unable to induce G2 arrest, was capable of inducing the degradation of APOBEC3H (Fig. 1C, lanes 1 and 2); conversely, VifNL4–3, which functions as a cell cycle regulator and causes G2 arrest, was unable to remove APOBEC3H through proteasomal degradation (Fig. 1C, lanes 3 and 4). As shown in Figures 1D and 1E, both VifHXB2 and VifNL4–3 have been repeatedly reported by various groups to be functional against APOBEC3F/G.5,10,15,17,46 Therefore, the observed distinction in functionality concerning APOBEC3H depletion and G2 arrest could not be attributed to differences in E3 ubiquitin complex formation; instead, it suggests that Vif may selectively choose one function over the other.
Figure 1.

Vifs that are competent for APOBEC3H removal cannot induce cell cycle arrest. (A) Representative flow cytometry data showing VifNL4–3's or VifHXB2's potency in cell cycle regulation in HEK293T cells. (B) Bar chart representing the (G2/M)/G1 ratio of HEK293T cells transfected with VifNL4–3 or VifHXB2. Western blotting results (above) indicate the protein expression levels of VifNL4–3 and VifHXB2. (C) Western blotting results for APOBEC3H levels in the presence of VifHXB2 or VifNL4–3 in HEK293T cells. (D) Western blotting results for APOBEC3G levels in the presence of VifHXB2 or VifNL4–3 in HEK293T cells. (E) Western blotting results for APOBEC3F levels in the presence of VifHXB2 or VifNL4–3 in HEK293T cells. (F) Bar chart representing the (G2/M)/G1 ratio of HEK293T cells transfected with different Vifs that are potent in depleting APOBEC3H, with the exclusion of BNL43. (G) Western blotting results indicating the protein expression levels of the Vif proteins in (F).
To further confirm that APOBEC3H depletion and G2 arrest are selective features among HIV-1 Vif proteins, we tested several Vif proteins that have been shown to be functional against APOBEC3H and belong to different HIV-1 subtypes.41 Unlike VifNL4–3, all HIV-1 Vif proteins that are known to induce the degradation of A3H were unable to arrest host cells at the G2/M stage (Fig. 1F), despite efficient expression in HEK293T cells (Fig. 1G). It is noteworthy that Vifs from NL4–3, LAI, and B1, all belonging to same subtype B, showed different potencies in arresting host cells at the G2/M stage, suggesting that cell cycle regulation of Vif protein may not correlate with HIV-1 subtype.
HIV-1 Vif proteins that are competent for inducing G2/M cell cycle arrest are defective for APOBEC3H depletion
The data presented above suggest that APOBEC3H depletion and G2 arrest are selective features that cannot co-exist on a single Vif molecule. To further examine this possibility, we characterized additional HIV-1 Vif proteins that are competent with regard to inducing G2 arrest. We observed that VifSG3 and VifC2 also had the ability to induce G2 arrest in HEK293T cells (Fig. 2A). In repeated experiments, VifSG3 and VifC2 demonstrated an ability to cause G2 arrest, like that of VifNL4–3 (Fig. 2B). However, all 3 of these Vifs were defective in inducing APOBEC3H degradation when compared to VifHXB2 (Fig. 2C). On the other hand, all 3 were able to induce APOBEC3G degradation (Fig. 2D), indicating that their ability to form CRL5 E3 complexes was unaffected.
Figure 2.
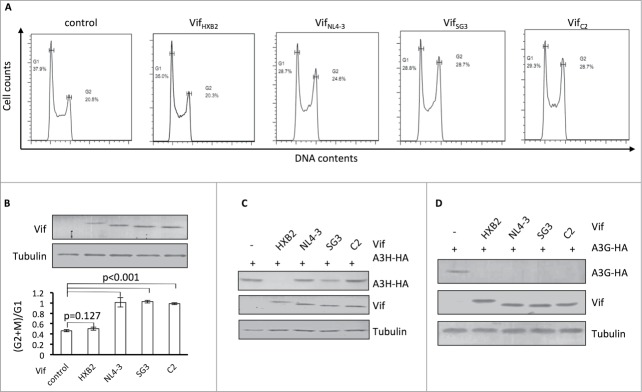
Vifs that are capable of G2 arrest cannot induce the depletion of APOBEC3H. (A) Representative flow cytometry data for cell cycle regulation by different Vifs in HEK293T cells. (B) Bar chart representing the (G2/M)/G1 ratios for HEK293T cells transfected with different Vif expression vectors. The Western blotting results (above) indicate the expression levels of Vif proteins. (C) Western blotting results for APOBEC3H levels in the presence of different Vif proteins in HEK293T cells. (D) Western blotting results for APOBEC3G levels in the presence of different Vif proteins in HEK293T cells
A single mutation at position 48 alters VifNL4–3's potency in causing G2 arrest and inability to cause APOBEC3H depletion
Residue 48 of Vif has been determined to be essential for Vif-induced APOBEC3H depletion.41,42 Since it is located between residues 47 and 50 in Vif proteins, altering residue 48 may also change Vif's potency in G2 arrest. To test this hypothesis, we focused on VifHXB2 and VifNL4–3, which indeed showed different potencies for the 2 functions (Fig. 1B, C). Position 48 has been shown to be critical for Vif-induced APOBEC3H degradation42 and is occupied by different residues in VifHXB2 (an H residue) and VifNL4–3 (an N, Fig. 3A). We have previously demonstrated that VifNL4–3 N48H, as opposed to wild-type VifNL4–3, promotes viral infectivity in the presence of antiviral APOBEC3H.42 In this study, we checked the effects of similar mutations on Vif's potency in cell cycle regulation. Mutation N48H completely abolished VifNL4–3's ability to arrest host cells at the G2/M stage (Fig. 3B), demonstrating that position 48 is indeed involved in Vif-induced G2 arrest. Combined with our previous observation, these data suggest that different residues at position 48 alter VifNL4–3s capacity for APOBEC3H depletion and G2 arrest. However, mutation H48N did not confer on VifHXB2 the ability to cause G2 arrest in host cells (Fig. 3B), suggesting that residue 48 alone is not sufficient for Vif-induced G2 arrest and that this process requires other specific residues of Vif proteins.
Figure 3.
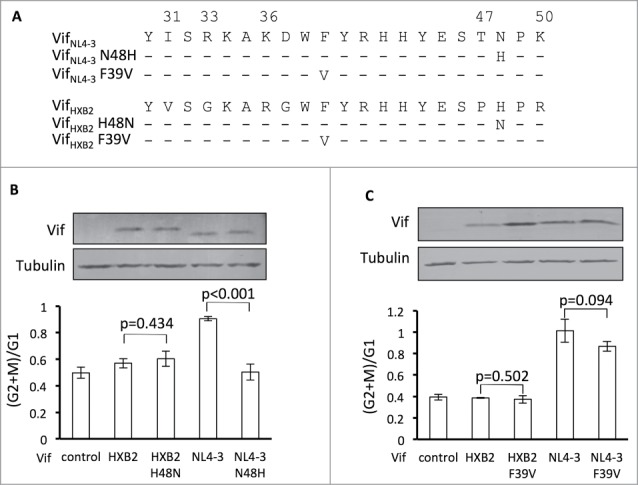
Position 48 is essential for Vif's induction of G2 arrest or depletion of APOBEC3H. (A) VifHXB2 and VifNL4–3 mutants used in this figure. (B) Bar chart representing the (G2/M)/G1 ratios in HEK293T cells transfected with VifHXB2, VifNL4–3, and their mutants at position 48. The Western blotting results (above) indicate the protein expression levels of VifNL4–3 and VifHXB2. (C) Bar chart representing the (G2/M)/G1 ratios in HEK293T cells transfected with VifHXB2, VifNL4–3, and their mutants at position 39. The Western blotting results (above) indicate the protein expression levels of VifNL4–3 and VifHXB2.
In situ, residue 39 of Vif is physically close to residues 31, 33, and 36 and is also critical for APOBEC3H depletion.41 VifNL4–3 and VifHXB2 share the same amino acid at residue 39. To test the impact of this residue on G2 arrest, we introduced an F39V mutation into both Vif molecules. To our surprise, F39V neither abolished the G2 arrest potency of VifNL4–3 nor made VifHXB2 capable of G2 arrest (Fig. 3C), possibly because residue 39 is not close enough to residues 31/33/36, and therefore the mutagenesis of F39 to V alone cannot affect Vif's function as a cell cycle regulator.
Identification of amino acids critical for Vif-induced G2 arrest
To determine the critical amino acids for Vif-induced G2 arrest, we performed a protein sequence alignment using G2 arrest-competent VifNL4–3, VifSG3, and VifC2, as well as G2 arrest-defective VifHXB2. Interestingly, the G2 arrest-competent Vif proteins share identical residues at positions 31, 33, 36, and 50, which are different from those in VifHXB2 (Fig. 4A). Positions 47 of VifNL4–3, VifSG3, and VifC2 also differ from that of VifHXB2, but VifC2 contains a different amino acid at position 47 than VifNL4–3 and VifSG3 (R instead of T; Fig. 4A).
Figure 4.
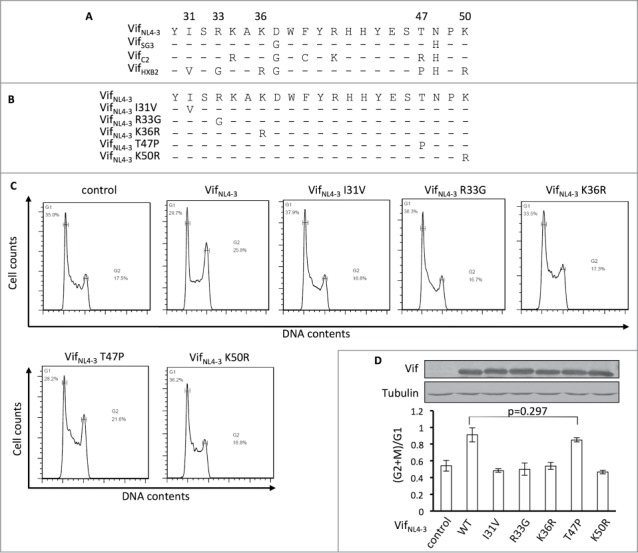
Positions 31, 33, 36, and 50, but not 47, are critical for Vif's induction of G2 arrest. (A) Protein alignment of G2-competent Vif proteins. G2-incompetent VifHXB2 was also introduced. The results suggest that position 47 is not conserved among different G2 arrest-competent Vifs. Numbers above the alignment indicate previously determined critical positions for Vif's induction of G2 arrest. Hyphens ("-") represent identical residues, as compared to VifNL4–3. (B) VifNL4–3 mutants used in this figure. (C) Representative flow cytometry data for cell cycle regulation by VifNL4–3 and its mutants. (D) Bar chart representing the (G2/M)/G1 ratios in HEK293T cells transfected with VifNL4–3 and its mutants. The Western blotting results (above) indicate protein expression levels of VifNL4–3.
To further determine whether all of these 5 residues are essential for Vif-induced G2 arrest, we individually substituted single mutations at positions 31, 33, 36, 47, and 50 in VifNL4–3 (Fig. 4B) and then tested for potency in arresting host cells at the G2/M stage. We found that single mutations I31V, R33G, K36R, and K50R were each individually sufficient to reduce or abolish VifNL4–3's ability to induce G2 arrest. Mutant T47P maintained the ability to cause G2 arrest (Fig. 4C, D), suggesting that this position is less important for determining the molecule's capacity for causing G2 arrest. Therefore, positions 31, 33, 36, and 50, but not 47, are apparently critical for Vif-induced G2 arrest.
Exchanging residues at positions 31, 33, 36, 47, and 50 alters the G2 arrest and APOBEC3H depletion capability of VifHXB2
Another interesting phenomenon revealed by the alignment of the Vif proteins was that VifSG3 and VifHXB2 contain identical residues at positions 39 and 48 (Fig. 4A); however, unlike VifHXB2, VifSG3 failed to deplete APOBEC3H (Fig. 2C). It is worth noting that the major difference between the N-termini (positions 1–80) of VifSG3 and VifHXB2 is at residues 31, 33, 36, 47, and 50 (Fig. 5A), and it has previously been reported that a VifHXB2 mutant containing VifNL4–3 residues at positions 31, 33, 36, 47, and 50 (VifHXB2 M5, Fig. 5A) shows potency in inducing host cell arrest at the G2/M stage.45 Since the regions involved in APOBEC3H depletion and G2 arrest now appear to overlap in the Vif protein, it is reasonable to conclude that altering residues at 31, 33, 36, 47, and 50 in VifHXB2 and VifNL4–3 (Fig. 5A) would affect their ability to deplete APOBEC3H. Indeed, further study indicated that mutations at positions 33, 36, or 47, or combinations of G2-related positions, compromised VifHXB2-induced APOBEC3H depletion (Fig. 5B); although VifHXB2 M5 was potent in causing G2 arrest, it almost completely lost its potency in inducing APOBEC3H degradation (Fig. 5C). Interestingly, VifNL4–3 M5 did not gain the same ability to remove APOBEC3H (Fig. 5C), possibly because of the negative effect of N48 on APOBEC3H depletion.
Figure 5.
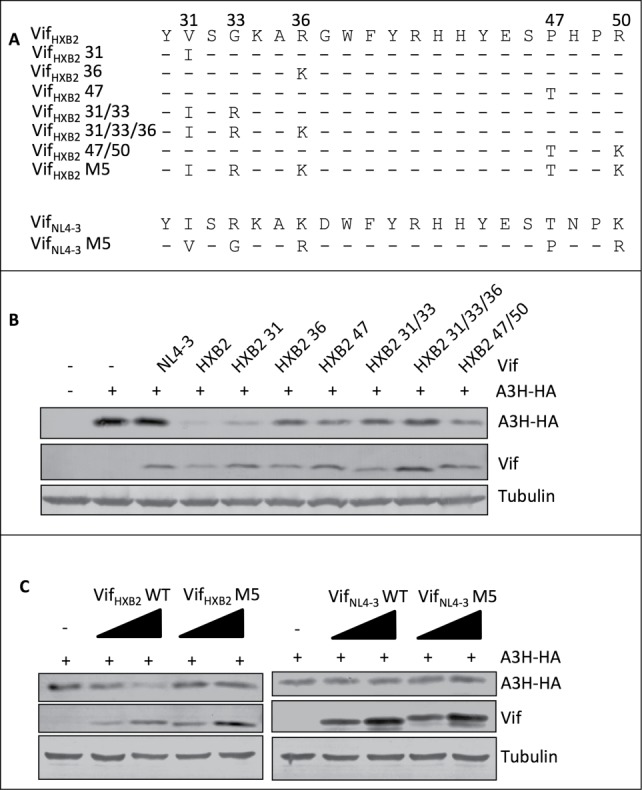
G2-related positions are also critical for Vif's induction of APOBEC3H depletion. (A) VifHXB2 and VifNL4–3 mutants used in this figure. (B) Western blotting results for APOBEC3H levels in the presence of VifHXB2 and its mutants at G2-related positions. VifNL4–3 was introduced as a negative control. (C) Western blotting results for APOBEC3H levels in the presence of VifHXB2, VifNL4–3, and their M5 mutants (with positions 31, 33, 36, 47, and 50 exchanged between VifHXB2 and VifNL4–3).
Critical residues that are responsible for APOBEC3H depletion and G2 arrest are located in similar areas of the Vif protein.
Recently, Guo et al. reported the structure of VifNL4–3,19 which revealed the reason why one Vif protein could induce the degradation of both APOBEC3F and APOBEC3G. Indeed, Vif uses 2 distinct surface areas to interact with and deplete APOBEC3F (Fig. 6A, residues in blue) or APOBEC3G (Fig. 6A, residues in wheat). The phenomenon we observed above, that Vif could not simultaneously function to deplete APOBEC3H and arrest host cells at the G2/M stage, led us to hypothesize that Vif uses the same area for both functions. Fortunately, Guo's Vif structure was based on VifNL4–3, which is competent for G2 arrest and thus provides conformational information concerning Vif-induced cell cycle regulation.
Figure 6.
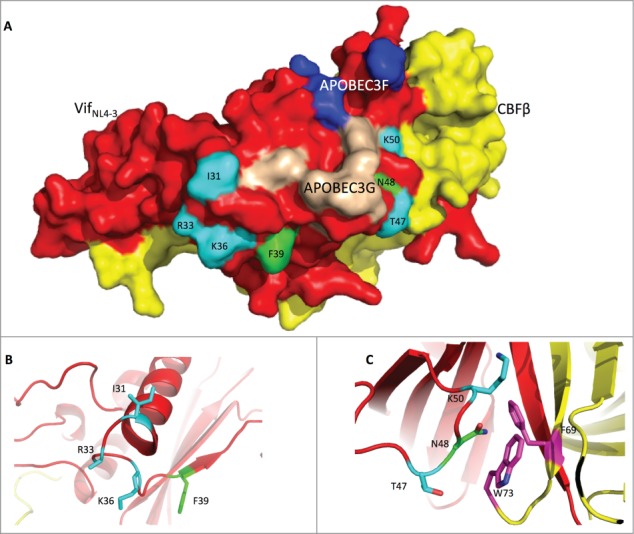
The Vif protein uses similar regions to induce G2 arrest and the degradation of APOBEC3H. (A) Surface model of the Vif-CBFβ interaction based on the published structure (PDB: 4N9F).19 The Vif protein is labeled in red, and CBFβ is in yellow. On the Vif protein, the APOBEC3F-binding region is labeled in blue, the APOBEC3G-binding region in wheat, the critical residues for G2 arrest in cyan, and the critical residues for APBOEC3H depletion in green. (B) Detailed conformation of Vif around positions 31, 33, 36 (essential for G2 arrest), and 39 (essential for APOBEC3H depletion). (C) Detailed conformation of Vif around positions 47, 50 (for G2 arrest), and 48 (for APBOEC3H depletion). It is noteworthy that N48 on Vif may interact with F69 and W73 (in purple) on CBFβ.
We first noted that residues F39 and N48, which are critical for APOBEC3H depletion, are found in 2 different areas in VifNL4–3 (Fig. 6A, residues in green). Residue F39 is located on one side of the Vif β-sheet, with its side-chain facing outwards (Fig. 6B); this configuration would be consistent with binding APOBEC3H. Shockingly, though, residue N48, on the other side, is barely on the surface of the Vif protein; instead, its side chain protrudes into CBFβ, and it interacts with this protein, possibly through F69 and W73 (residues in purple) of CBFβ (Fig. 6C), suggesting that APOBEC3H depletion requires a proper interaction between Vif and CBFβ.
Interestingly, consistent with our hypothesis, residues that are important for G2 arrest are also located in these 2 distinct areas (Fig. 6A, residues in cyan). Residues I31, R33, and K36 are very close to F39 (Fig. 3B). With their side-chains facing outwards, these residues are poised to interact with the unknown endogenous cell cycle regulator, so that Vif could deplete the regulator and cause G2 arrest. Residues T47 and K50 are located beside N48 (Fig. 6C). Like N48, these 2 residues are also very close to CBFβ, indicating that G2 arrest probably also needs a very specific interaction between Vif and CBFβ. Thus, residues critical for APOBEC3H depletion and G2 arrest are located in 2 similar areas of Vif.
Discussion
In addition to depleting the antiviral factors APOBEC3G and APOBEC3F, the HIV-1 Vif protein can also increase viral infectivity through APOBEC3H degradation37,39,46,47 or increase viral production through G2 arrest.45 Unlike APOBEC3F/G, APOBEC3H has several haplotypes, most of which are unstable and incompetent in HIV-1 suppression.37 Thus, it is possible that only antiviral-competent APOBEC3H can exert additional selection pressure on HIV-1. Consistently, only some Vifs from different HIV-1 strains show potency in depleting APOBEC3H,41 and we have further confirmed that HIV-1 Vif adapts to APOBEC3H haplotypes in HIV-1-infected patients.40
Based on previous observations with VifNL4–3, VifHXB2, and VifSG3, G2 arrest is another feature that is selectively found among these HIV-1 Vif proteins. Interestingly, screening of additional Vifs from different HIV-1 strains/subtypes has further confirmed that APOBEC3H and G2 arrest cannot co-exist on a single Vif protein. Consistently, based on the reported structure,19 critical residues in VifNL4–3 such as positions 39 and 48 for APOBEC3H depletion and 31, 33, 36, 47, and 50 for G2 arrest span the same surface on the Vif protein. Interestingly, changing residue 48, which is critical for Vif-induced APOBEC3H depletion, alters VifNL4–3's potency in G2 arrest. On the other hand, VifSG3 with a phenylalanine at position 39 and histidine at position 48 cannot potently reduce APOBEC3H, even though both F39 and H48 are necessary for APOBEC3H removal; consistently, VifHXB2 M5 with F39 and H48 showed almost no ability to reduce APOBEC3H. Further research has demonstrated that mutations in G2-related positions, such as 33, 36, and 47, compromise VifHXB2's capacity to counteract APOBEC3H to varying degrees, while altering all 5 G2-associated residues together may lead to a more significant change on Vif's surface and cause greater differences in the recognition of APOBEC3H. Thus, taken together, these data not only establish that these G2-related positions are important for Vif-mediated depletion of APOBEC3H, but they again confirm that a single Vif protein can only be capable of either APOBEC3H depletion or G2 arrest.
The overlap of critical residues for APOBEC3H depletion and G2 arrest makes it reasonable for Vif to have to choose one function over the other. In fact, without the presence of antiviral APOBEC3H but with other selection pressure(s), it is reasonable to conclude that HIV-1 alters residues critical for APOBEC3H degradation and improves the infectivity of the produced viruses through additional mechanism(s). Because of the very same reason and the constitutive presence of antiviral-competent APBOEC3F/G in HIV-1 target cells, regions of Vif that are directed against APOBEC3F/G are highly conserved among different Vifs. Although their contribution to HIV-1 pathogenesis is still unclear, it is at least known that Vif-induced G2 arrest elevates HIV-1 production45 and therefore should promote the development of AIDS. However, the selection pressure connected with G2 arrest is unknown. With only a few Vif proteins being capable of delaying the cell cycle,44,48,49 it is most likely that this function has arisen as a result of high-level random mutagenesis of the HIV-1 genome (because of the relatively low fidelity of reverse transcription, and/or mutagenesis of APOBEC3-mediated RNA editing), and elevated viral production subsequently makes it advantageous for these particular HIV-1 strains to retain this ability to delay the cell cycle during viral replication. Thus, Vif proteins with no potency in either function may represent intermediates in the mutagenesis. Otherwise, Vif's capacity to regulate the cell cycle may be evolutionarily increased under certain selective pressures, and Vifs that are incompetent with regard to either APOBEC3H depletion or G2 arrest must be indicatative of additional selective function(s) that we have not yet discovered for Vif.
Cellular factor CBFβ has proven critical for HIV-1 Vif function,15,16,18,20,21,50,51 since it facilitates the interaction of Vif with Cul5, the core protein of the Vif-induced E3 complex.5 We have also determined that CBFβ is a key factor in Vif-induced G2 arrest (unpublished data, manuscript submitted), which correlates with previous discovery that Vif-induced G2 arrest is Cul5-dependent.44,48,49 Interestingly, no Vif residue that has been shown to be critical for APOBEC3F/G depletion has been found to be located at the Vif-CBFβ binding interface (except for those residues compromising Vif E3 complex formation). In our study, however, Vif mutations at the Vif-CBFβ interface (such as Vif residues 48 and 50) alter its potency in APOBEC3H depletion and G2 arrest. VifHXB2 and VifNL4–3, while containing such different residues, maintain similar activity in APOBEC3F/G degradation,42,45 indicating that amino acid differences at positions 48 and 50 do not significantly alter Vif's structure or the assembly of the Vif-induced E3 complex. However, since VifHXB2 H48N can no longer induce the degradation of APOBEC3H, and VifNL4–3 K50R cannot cause G2 arrest, our findings not only again confirm the involvement of CBFβ in Vif-induced G2 arrest but also indicate that Vif requires some specific interaction with CBFβ to induce the degradation of APOBEC3H or to arrest host cells at G2.
Materials and Methods
Plasmids
The construction of expression vectors for Vifs from different HIV-1 subtypes has been previously described,41 as have expression vectors for CBFβ and its mutants.15,20 Mutations were introduced into Vif by site-directed mutagenesis, with TransStart KD Plus DNA Polymerase (Transgen Biotech, AP301–11) and the following primers: VifNL4–3 I31V forward: 5′-AAGCACCACATGTACGTTAGCCGCAAAGC-3′; VifNL4–3 I31V reverse: 5′-GCTTTGCGGCTAACGTACATGTGGTGCTT-3′; VifNL4–3 R33G forward: 5′-CCACATGTACATTAGCGGCAAAGCTAAGGACTGG-3′; VifNL4–3 R33G reverse: 5′-CCAGTCCTTAGCTTTGCCGCTAATGTACATGTGG-3′; VifNL4–3 K36R forward: 5′-GCCGCAAAGCTAGGGACTGGTTCTACCG-3′; VifNL4–3 K36R reverse: 5′-CGGTAGAACCAGTCCCTAGCTTTGCGGC-3′; VifNL4–3 T47P forward: 5′-ACCACTACGAGAGCCCCAACCCCAAGATTAG-3′; VifNL4–3 T47P reverse: 5′-CTAATCTTGGGGTTGGGGCTCTCGTAGTGGT-3′; VifNL4–3
K50R forward: 5′-AGCACCAACCCCAGGATTAGCAGCGAGG-3′; VifNL4–3 K50R reverse: 5′-CCTCGCTGCTAATCCTGGGGTTGGTGCT-3′; VifNL4–3 F39V forward: 5′-AAGCTAAGGACTGGGTGTACCGCCACCAC-3′; VifNL4–3 F39V reverse: 5′- GTGGTGGCGGTACACCCAGTCCTTAGCTT-3′; VifNL4–3 I31V/R33G forward: 5′- CCACATGTACGTTAGCGGCAAAGCTAAGGACTGG-3′; VifNL4–3 I31V/R33G reverse: 5′-CCAGTCCTTAGCTTTGCCGCTAACGTACATGTGG-3′; VifNL4–3 I31V/R33G/K36R forward: 5′-GCGGCAAAGCTAGGGACTGGTTCTACCG-3′; VifNL4–3 I31V/R33G/K36R reverse: 5′-CGGTAGAACCAGTCCCTAGCTTTGCCGC-3′; VifNL4–3 T47P/K50R forward: 5′-AGCCCCAACCCCAGGATTAGCAGCGAGG-3′; VifNL4–3 T47P/K50R reverse: 5′-CCTCGCTGCTAATCCTGGGGTTGGGGCT-3′; VifNL4–3 N48H forward: 5′- ACTACGAGAGCACCCACCCCAAGATTAGCA-3′; VifNL4–3 N48H reverse: 5′- TGCTAATCTTGGGGTGGGTGCTCTCGTAGT-3′; VifHXB2 V31I forward: 5′- GTAAAACACCATATGTATATTTCAGGGAAAGCT-3′; VifHXB2 V31I reverse: 5′- AGCTTTCCCTGAAATATACATATGGTGTTTTAC-3′; VifHXB2 R36K forward: 5′- TCAAGGAAAGCTAAGGGATGGTTTTAT-3′; VifHXB2 R36K reverse: 5′- ATAAAACCATCCCTTAGCTTTCCTTGA-3′; VifHXB2 P47T forward: 5′- GACATCACTATGAAAGCACTCATCCAAGAATAAGT-3′; VifHXB2 P47T reverse: 5′- ACTTATTCTTGGATGAGTGCTTTCATAGTGATGTC-3′; VifHXB2 V31I/G33R forward: 5′-GTAAAACACCATATGTATATTTCAAGGAAAGCTAGGGG-3′; VifHXB2 V31I/G33R reverse: 5′-CCCCTAGCTTTCCTTGAAATATACATATGGTGTTTTAC-3′; VifHXB2 P47T/R50K forward: 5′-CATCACTATGAAAGCACTCATCCAAAGATAAGTTCAGAAGTACA-3′; VifHXB2 P47T/R50K reverse: 5′-TGTACTTCTGAACTTATCTTTGGATGAGTGCTTTCATAGTGATG-3′; VifHXB2 F39V forward: 5′- GCTAGGGGATGGGTGTATAGACATCACTATGAA-3′; VifHXB2 F39V reverse: 5′- TTCATAGTGATGTCTATACACCCATCCCCTAGC-3′; VifHXB2 H48N forward: 5′- TATGAAAGCCCTAATCCAAGAATAAGTTC-3′; VifHXB2 H48N reverse: 5′- GAACTTATTCTTGGATTAGGGCTTTCATA-3′. All transfections were performed with the help of PEI (Polysciences, 408727–100 ML), according to the manufacturer's protocol.
Antibodies
The following primary and secondary antibodies were used for this study: primary antibodies anti-HA (Invitrogen, 715500), anti-myc (Covance, MMS-101P), anti-V5 (Invitrogen, R960-CUS) and anti-tubulin (Abcam, ab11323–100); secondary antibodies anti-mouse (Jackson, 115–055–062) and anti-rabbit (Jackson, 111–055–045). Anti-Vif (catalog no. 2221) antibody was obtained from the AIDS Research and Reference Reagents Program.
Vif-induced G2 arrest
To detect Vif-induced G2 arrest, human embryo kidney 293T (HEK293T) cells seeded into 6-well plates were co-transfected with the pEGFP-F plasmid49 and one of the Vif-expressing vectors. At 48 h post-transfection, the transfected cells were harvested and subjected to propidium iodide (PI) staining and flow cytometry: The harvested cells were first washed twice with ice-cold PBS, then fixed in ice-cold ethanol at 4°C overnight. Cells were then washed in PBS twice and gently suspended in PI solution (0.1% Triton X-100, 5 mM EDTA, 50 μg/ml RNase A, and 50∼100 μg/ml PI [Sigma, P4170]), then incubated in the dark on ice for 1 h before flow cytometry. Stained cells were examined on a FACS Calibur flow cytometer (BD) with gating criteria similar those previously described.44 The flow cytometry data were then analyzed with FLOWJO software (Version 7.6.1).
Vif-induced protein depletion
To detect Vif-induced degradation of APOBEC3H or APOBEC3G, HEK293T cells seeded into 12-well plates were co-transfected with an APOBEC3H (Hap II)- or APOBEC3G-expressing vector, and one of the Vif-expressing vectors. At 48 h post-transfection, transfected cells were harvested and washed with 1× PBS. The harvested cells were then resuspended in 1× SDS-PAGE loading buffer and heated at 95°C for 10 min for SDS-PAGE, followed by transfer to nitrocellulose membranes (GE Whatman, 10401396). After blocking with PBS–Tween 20 containing 5% bovine serum albumin for 1 h at room temperature, the blots were incubated with a specific antibody overnight at 4°C. After three washes, the blots were stained with an alkaline phosphatase-conjugated secondary antibody for 1 h at room temperature. After three washes with PBS–Tween 20, the blots were reacted with nitroblue tetrazolium (NBT) (Sigma, N6876) and 5-bromo-4-chloro-3-indolylphosphate (BCIP) (Roche, 11585002001).
Protein modeling
The structure of the VifNL4–3-CBFβ interaction was based on the published structure of the Vif-induced E3 complex19 (PDB: 4N9F). The modeling and color manipulation were performed with PyMOL software (http://www.pymol.org, version 1.6.9.0).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. J. Wang for critical reagents; P. Li and H. Wang for technical assistance; R. Markham, J. Margolick, and J. Bream for thoughtful discussions; and D. McClellan for editorial assistance.
Funding
This work was supported in part by funding from the Chinese Ministry of Science and Technology (2012CB911100 and No. 2013ZX0001–005) and Chinese Ministry of Education (IRT1016), the Key Laboratory of Molecular Virology, Jilin Province (20102209), China, National Natural Science Foundation (81401654) to JD, a grant (470110000002) from Norman Bethune Health Science Center of Jilin University, a grant (R01 AI089246) from NIH to VAS, and a grant (2R56AI62644–6) from the NIAID.
Author Contribution
KZ, JD, YR, WZ (Zheng), JK, and JH performed experiments. KZ, JD, KW, WZ (Zhang), VAS and X-FY analyzed the data. JD and X-FY directed the project and wrote the paper with help from all authors.
References
- 1. Fisher AG, Ensoli B, Ivanoff L, Chamberlain M, Petteway S, Ratner L, Gallo RC, Wong-Staal F. The sor gene of HIV-1 is required for efficient virus transmission in vitro. Science 1987; 237:888-93; PMID:3497453; http://dx.doi.org/ 10.1126/science.3497453 [DOI] [PubMed] [Google Scholar]
- 2. Strebel K, Daugherty D, Clouse K, Cohen D, Folks T, Martin MA. The HIV A (sor) gene product is essential for virus infectivity. Nature 1987; 328:728-30; PMID:2441266; http://dx.doi.org/ 10.1038/328728a0 [DOI] [PubMed] [Google Scholar]
- 3. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002; 418:646-50; PMID:12167863; http://dx.doi.org/ 10.1038/nature00939 [DOI] [PubMed] [Google Scholar]
- 4. Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 2003; 114:21-31; PMID:12859895; http://dx.doi.org/ 10.1016/S0092-8674(03)00515-4 [DOI] [PubMed] [Google Scholar]
- 5. Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003; 302:1056-60; PMID:14564014; http://dx.doi.org/ 10.1126/science.1089591 [DOI] [PubMed] [Google Scholar]
- 6. Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med 2003; 9:1398-403; PMID:14528301; http://dx.doi.org/ 10.1038/nm946 [DOI] [PubMed] [Google Scholar]
- 7. Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med 2003; 9:1404-7; PMID:14528300; http://dx.doi.org/ 10.1038/nm945 [DOI] [PubMed] [Google Scholar]
- 8. Liu B, Yu X, Luo K, Yu Y, Yu XF. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J Virol 2004; 78:2072-81; PMID:14747572; http://dx.doi.org/ 10.1128/JVI.78.4.2072-2081.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J Biol Chem 2005; 280:18573-8; PMID:15781449; http://dx.doi.org/ 10.1074/jbc.C500082200 [DOI] [PubMed] [Google Scholar]
- 10. Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathogens 2005; 1:e6; PMID:16201018; http://dx.doi.org/ 10.1371/journal.ppat.0010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shirakawa K, Takaori-Kondo A, Kobayashi M, Tomonaga M, Izumi T, Fukunaga K, Sasada A, Abudu A, Miyauchi Y, Akari H, et al. . Ubiquitination of APOBEC3 proteins by the Vif-Cullin5-ElonginB-ElonginC complex. Virology 2006; 344:263-6; PMID:16303161; http://dx.doi.org/ 10.1016/j.virol.2005.10.028 [DOI] [PubMed] [Google Scholar]
- 12. Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev 2004; 18:2867-72; PMID:15574593; http://dx.doi.org/ 10.1101/gad.1250204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev 2004; 18:2861-6; PMID:15574592; http://dx.doi.org/ 10.1101/gad.1249904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sakai H, Shibata R, Sakuragi J, Sakuragi S, Kawamura M, Adachi A. Cell-dependent requirement of human immunodeficiency virus type 1 Vif protein for maturation of virus particles. J Virol 1993; 67:1663-6; PMID:8437236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang W, Du J, Evans SL, Yu Y, Yu XF. T-cell differentiation factor CBF-β regulates HIV-1 Vif-mediated evasion of host restriction. Nature 2012; 481:376-9 [DOI] [PubMed] [Google Scholar]
- 16. Han X, Liang W, Hua D, Zhou X, Du J, Evans SL, Gao Q, Wang H, Viqueira R, Wei W, et al. . Evolutionarily conserved requirement for core binding factor β in the assembly of the human immunodeficiency virus/simian immunodeficiency virus Vif-cullin 5-RING E3 ubiquitin ligase. J Virol 2014; 88:3320-8; PMID:24390335; http://dx.doi.org/ 10.1128/JVI.03833-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou X, Evans SL, Han X, Liu Y, Yu XF. Characterization of the interaction of full-length HIV-1 Vif protein with its key regulator CBFbeta and CRL5 E3 ubiquitin ligase components. PloS One 2012; 7:e33495; PMID:22479405; http://dx.doi.org/ 10.1371/journal.pone.0033495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou X, Han X, Zhao K, Du J, Evans SL, Wang H, Li P, Zheng W, Rui Y, Kang J, et al. . Dispersed and Conserved Hydrophobic Residues of HIV-1 Vif Are Essential for CBFbeta Recruitment and A3G Suppression. J Virol 2014; 88:2555-63; PMID:24352440; http://dx.doi.org/ 10.1128/JVI.03604-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo Y, Dong L, Qiu X, Wang Y, Zhang B, Liu H, Yu Y, Zang Y, Yang M, Huang Z. Structural basis for hijacking CBF-β and CUL5 E3 ligase complex by HIV-1 Vif. Nature 2014; 505:229-33; PMID:24402281; http://dx.doi.org/ 10.1038/nature12884 [DOI] [PubMed] [Google Scholar]
- 20. Du J, Zhao K, Rui Y, Li P, Zhou X, Zhang W, Yu XF. Differential requirements for HIV-1 Vif-mediated APOBEC3G degradation and RUNX1-mediated transcription by core binding factor β. J Virol 2013; 87:1906-11; PMID:23175372; http://dx.doi.org/ 10.1128/JVI.02199-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hultquist JF, McDougle RM, Anderson BD, Harris RS. HIV type 1 viral infectivity factor and the RUNX transcription factors interact with core binding factor β on genetically distinct surfaces. AIDS Res Hum Retroviruses 2012; 28:1543-51; PMID:22725134; http://dx.doi.org/ 10.1089/aid.2012.0142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang H, Liu B, Liu X, Li Z, Yu X-F, Zhang W. Identification of HIV-1 Vif Regions Required for CBF-β Interaction and APOBEC3 Suppression. PloS One 2014; 9:e95738; PMID:24810617; http://dx.doi.org/ 10.1371/journal.pone.0095738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci U S A 2005; 102:11444-9; PMID:16076960; http://dx.doi.org/ 10.1073/pnas.0502440102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xiao Z, Ehrlich E, Yu Y, Luo K, Wang T, Tian C, Yu XF. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology 2006; 349:290-9; PMID:16530799; http://dx.doi.org/ 10.1016/j.virol.2006.02.002 [DOI] [PubMed] [Google Scholar]
- 25. Mehle A, Thomas ER, Rajendran KS, Gabuzda D. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J Biol Chem 2006; 281:17259-65; PMID:16636053; http://dx.doi.org/ 10.1074/jbc.M602413200 [DOI] [PubMed] [Google Scholar]
- 26. Paul I, Cui J, Maynard EL. Zinc binding to the HCCH motif of HIV-1 virion infectivity factor induces a conformational change that mediates protein-protein interactions. Proc Natl Acad Sci U S A 2006; 103:18475-80; PMID:17132731; http://dx.doi.org/ 10.1073/pnas.0604150103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xiao Z, Ehrlich E, Luo K, Xiong Y, Yu XF. Zinc chelation inhibits HIV Vif activity and liberates antiviral function of the cytidine deaminase APOBEC3G. FASEB J 2007; 21:217-22; PMID:17135358; http://dx.doi.org/ 10.1096/fj.06-6773com [DOI] [PubMed] [Google Scholar]
- 28. Xiao Z, Xiong Y, Zhang W, Tan L, Ehrlich E, Guo D, Yu XF. Characterization of a novel Cullin5 binding domain in HIV-1 Vif. J Mol Biol 2007; 373:541-50; PMID:17869271; http://dx.doi.org/ 10.1016/j.jmb.2007.07.029 [DOI] [PubMed] [Google Scholar]
- 29. Evans SL, Schon A, Gao Q, Han X, Zhou X, Freire E, Yu XF. HIV-1 Vif N-terminal Motif is required for recruitment of Cul5 to Suppress APOBEC3. Retrovirology 2014; 11:4; PMID:24422669; http://dx.doi.org/ 10.1186/1742-4690-11-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Giri K, Maynard EL. Conformational analysis of a peptide approximating the HCCH motif in HIV-1 Vif. Biopolymers 2009; 92:417-25; PMID:19382167; http://dx.doi.org/ 10.1002/bip.21209 [DOI] [PubMed] [Google Scholar]
- 31. Giri K, Scott RA, Maynard EL. Molecular structure and biochemical properties of the HCCH-Zn2+ site in HIV-1 Vif. Biochemistry 2009; 48:7969-78; PMID:19588889; http://dx.doi.org/ 10.1021/bi900677w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He Z, Zhang W, Chen G, Xu R, Yu XF. Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J Mol Biol 2008; 381:1000-11; PMID:18619467; http://dx.doi.org/ 10.1016/j.jmb.2008.06.061 [DOI] [PubMed] [Google Scholar]
- 33. Pery E, Rajendran KS, Brazier AJ, Gabuzda D. Regulation of APOBEC3 proteins by a novel YXXL motif in human immunodeficiency virus type 1 Vif and simian immunodeficiency virus SIVagm Vif. J Virol 2009; 83:2374-81; PMID:19109396; http://dx.doi.org/ 10.1128/JVI.01898-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen G, He Z, Wang T, Xu R, Yu XF. A patch of positively charged amino acids surrounding the human immunodeficiency virus type 1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J Virol 2009; 83:8674-82; PMID:19535450; http://dx.doi.org/ 10.1128/JVI.00653-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tian C, Yu X, Zhang W, Wang T, Xu R, Yu XF. Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J Virol 2006; 80:3112-5; PMID:16501124; http://dx.doi.org/ 10.1128/JVI.80.6.3112-3115.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Russell RA, Pathak VK. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol 2007; 81:8201-10; PMID:17522216; http://dx.doi.org/ 10.1128/JVI.00395-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. OhAinle M, Kerns JA, Li MM, Malik HS, Emerman M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 2008; 4:249-59; PMID:18779051; http://dx.doi.org/ 10.1016/j.chom.2008.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harari A, Ooms M, Mulder LC, Simon V. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J virol 2009; 83:295-303; PMID:18945781; http://dx.doi.org/ 10.1128/JVI.01665-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tan L, Sarkis PT, Wang T, Tian C, Yu XF. Sole copy of Z2-type human cytidine deaminase APOBEC3H has inhibitory activity against retrotransposons and HIV-1. FASEB J 2009; 23:279-87; PMID:18827027; http://dx.doi.org/ 10.1096/fj.07-088781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ooms M, Brayton B, Letko M, Maio SM, Pilcher CD, Hecht FM, Barbour JD, Simon V. HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe 2013; 14:411-21; PMID:24139399; http://dx.doi.org/ 10.1016/j.chom.2013.09.006 [DOI] [PubMed] [Google Scholar]
- 41. Binka M, Ooms M, Steward M, Simon V. The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J Virol 2012; 86:49-59; PMID:22013041; http://dx.doi.org/ 10.1128/JVI.06082-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ooms M, Letko M, Binka M, Simon V. The resistance of human APOBEC3H to HIV-1 NL4-3 molcular clone is determined by a single amino acid in Vif. PloS One 2013; 8:e57744; PMID:23469063; http://dx.doi.org/ 10.1371/journal.pone.0057744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sakai K, Dimas J, Lenardo MJ. The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest. Proc Natl Acad Sci 2006; 103:3369-74; http://dx.doi.org/ 10.1073/pnas.0509417103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang J, Shackelford JM, Casella CR, Shivers DK, Rapaport EL, Liu B, Yu XF, Finkel TH. The Vif accessory protein alters the cell cycle of human immunodeficiency virus type 1 infected cells. Virology 2007; 359:243-52; PMID:17056089; http://dx.doi.org/ 10.1016/j.virol.2006.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Izumi T, Io K, Matsui M, Shirakawa K, Shinohara M, Nagai Y, Kawahara M, Kobayashi M, Kondoh H, Misawa N, et al. . HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc Natl Acad Sci U S A 2010; 107:20798-803; PMID:21071676; http://dx.doi.org/ 10.1073/pnas.1008076107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhen A, Wang T, Zhao K, Xiong Y, Yu XF. A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J Virol 2010; 84:1902-11; PMID:19939923; http://dx.doi.org/ 10.1128/JVI.01509-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhen A, Du J, Zhou X, Xiong Y, Yu XF. Reduced APOBEC3H variant anti-viral activities are associated with altered RNA binding activities. PloS One 2012; 7:e38771; PMID:22859935; http://dx.doi.org/ 10.1371/journal.pone.0038771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang J, Reuschel EL, Shackelford JM, Jeang L, Shivers DK, Diehl JA, Yu XF, Finkel TH. HIV-1 Vif promotes the G(1)- to S-phase cell-cycle transition. Blood 2011; 117:1260-9; PMID:21149631; http://dx.doi.org/ 10.1182/blood-2010-06-289215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang J, Shackelford JM, Selliah N, Shivers DK, O'Neill E, Garcia JV, Muthumani K, Weiner D, Yu X-F, Gabuzda D, et al. . The HIV-1 Vif Protein Mediates Degradation of Vpr and Reduces Vpr-Induced Cell Cycle Arrest. DNA Cell Biol 2008; 27:267-77; PMID:18462066; http://dx.doi.org/ 10.1089/dna.2007.0707 [DOI] [PubMed] [Google Scholar]
- 50. Jager S, Kim DY, Hultquist JF, Shindo K, Larue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, et al. . Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature 2012; 481:371-5; PMID:22190037; http://dx.doi.org/ 10.1038/nature10693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hultquist JF, Binka M, Larue RS, Simon V, Harris RS. Vif proteins of human and simian immunodeficiency viruses require cellular CBFbeta to degrade APOBEC3 restriction factors. J Virol 2012; 86:2874-7; PMID:22205746; http://dx.doi.org/ 10.1128/JVI.06950-11 [DOI] [PMC free article] [PubMed] [Google Scholar]