

Abstract
Beside its central role in the mitochondria-dependent cell death pathway, the apoptotic protease activating factor 1 (Apaf-1) is involved in the DNA damage response through cell-cycle arrest induced by genotoxic stress. This non-apoptotic function requires a nuclear translocation of Apaf-1 during the G1-to-S transition. However, the mechanisms that trigger the nuclear accumulation of Apaf-1 upon DNA damage remain to be investigated. Here we show that the main 4 isoforms of Apaf-1 can undergo nuclear translocation and restore Apaf-1 deficient MEFs cell cycle arrest in the S phase following genotoxic stress through activation of Chk-1. Interestingly, DNA damage-dependent nuclear accumulation of Apaf-1 occurs independently of p53 and the retinoblastoma (pRb) pathway. We demonstrated that Apaf-1 associates with the nucleoporin Nup107 and this association is necessary for Apaf-1 nuclear import. The CED-4 domain of Apaf-1 directly binds to the central domain of Nup107 in an ATR-regulated, phosphorylation-dependent manner. Interestingly, expression of the Apaf-1-interacting domain of Nup107 interfered with Apaf-1 nuclear translocation upon genotoxic stress, resulting in a marked reduction of Chk-1 activation and cell cycle arrest. Thus, our results confirm the crucial role of Apaf-1 nuclear relocalization in mediating cell-cycle arrest induced by genotoxic stress and implicate Nup107 as a critical regulator of the DNA damage-induced intra-S phase checkpoint response.
Keywords: Apoptosome, Apaf-1, cell cycle, cisplatin, DNA damage, nucleoporin, nuclear pore complex, Nup107
Abbreviations
- Apaf-1
apoptotic protease activating factor 1
- CARD
caspase activation recruitment domain
- NSCLC
non-small cell lung cancer
- MEFs
Mouse Embryonic Fibroblasts
- GST
glutathione S-transferase
- DAPI
4′-diamino-2-phenylindole
- PI
propidium iodide
Introduction
Apoptosis, an evolutionarily conserved cell suicide process, relies on the activation of caspases, a group of cysteine proteases which cleave an array of specific substrates to produce the characteristic morphological features of the apoptotic cell death.1,2 Caspase activation can be initiated by 2 main pathways, extrinsic and intrinsic. In vertebrates, the intrinsic pathway requires the release of cytochrome c from mitochondria, which then interacts with the CED-4 homolog Apaf-1.3 In the presence of dATP/ATP, cytochrome c binding triggers the oligomerization of Apaf-1 into a caspase-activating complex, the apoptosome, which sequentially recruits and activates the initiator caspase-9.4,5 Activated caspase-9 in turn cleaves and activates downstream caspases including caspase-3 and caspase-7. Apaf-1 is a multidomain adaptor protein comprised of an N-terminal caspase recruitement domain (CARD), followed by a nucleotide binding/oligomerization domain that is homologous to CED-4 and a series of 12–13 C-terminal WD40 repeats. Multiple Apaf-1 splice variants can exist, but not all isoforms thus produced can activate procaspase-9.6,7 Indeed, in tumor cell lines, alternative splicing can create 4 main isoforms of Apaf-1, which can be distinguished by the presence or absence of an N-terminal 11 amino acid insert between the CARD and the CED-4 domains or an additional C-terminal WD40 between the fifth and the sixth WD40s. Only those isoforms with the additional WD40 repeat can efficiently associate with cytochrome c and activate caspase-9.6 Beside its role in the activation of caspase-9, nonapoptotic functions of Apaf-1 have been discovered.8,9 Among those, Apaf-1 has been demonstrated to be involved in the DNA damage response in mediating cell-cycle arrest induced by genotoxic stress.9 Indeed, Apaf-1 knockdown in human cancer cells reduced the activating phosphorylation of Chk1 following genotoxic stress, such as sublethal doses of cisplatin, which compromised the S phase arrest of treated cells.9 Interestingly, this cell-cycle-related function of Apaf-1 was not modulated by caspases inhibitors and occurred in cells treated with low doses of cisplatin that were not sufficient to induce apoptosis, indicating that the influence of Apaf-1 on the cell cycle is independent of its apoptotic role.9 Whereas Apaf-1 mostly resides in the cytoplasm of healthy cells,10 DNA damage elicits a rapid nuclear translocation of Apaf-1, independently from the apoptosis-related nuclear permeabilization.9,11,12 This nuclear translocation of Apaf-1, which seems to be regulated by the ataxia-telangiectasia-mutated (ATM) and the ATM- and Rad3-related (ATR) kinases, precedes the activation of checkpoint kinase-1 (Chk1), suggesting that Apaf-1 relocalization is critically involved in the ATR/Chk1 pathway activated by DNA damage.9 Interestingly, the nuclear presence of Apaf-1 constitutes a positive prognostic in non-small cell lung cancer (NSCLC) patients.9,13 However, the mechanisms that trigger the nuclear accumulation of Apaf-1 upon DNA damage remain to be determined. Here we investigated the putative role of the main Apaf-1 isoforms in the regulation of cell cycle. We show that the studied 4 isoforms of Apaf-1 can undergo nuclear translocation and complement the partial reduction of Chk1 activating phosphorylation in Apaf-1 deficient MEFs upon DNA damage, thus restoring genotoxic stress-dependent cell cycle arrest. Apaf-1 is imported to the nucleus by a p53- and pRb-independent mechanism involving direct binding to the nucleoporin Nup107 that is favored by ATR-regulated phosphorylation of Apaf-1. These data confirm that nuclear import of Apaf-1 is necessary for genotoxic stress-induced cell-cycle arrest and implicate the nucleoporin Nup107 as a regulator of the DNA damage response.
Results
Apaf-1 variants translocate to the nucleus and elicit cell-cycle arrest in Apaf-1-deficient MEFs upon DNA damage
Multiple splice variants of Apaf-1 have been described, which are known to have different abilities to activate caspase-9. To examine the role of the Apaf-1 variants the DNA damage response, we transduced the 4 main forms of Apaf-1 (Fig. 1A) into apaf-1−/− MEFs and studied their respective subcellular distribution as well as their cell cycle regulatory effect in the absence and following genotoxic stress. The expression levels of the different studied forms of Apaf-1 in the obtained cell lines were comparable to that of endogenous Apaf-1 in WT MEFs (Fig. 1B). Consistent with previous findings,6 comparative analysis indicated that only the isoforms containing the extra WD40 repeat were able of cytochrome c-dependent activation of procaspase-9 (Fig. 1C). As described previously,9 treatment of wild-type (WT) MEFs with a low, subapoptotic dose (20 μM) of cisplatin induces translocation of Apaf-1 from the cytoplasm to the nucleus, as well as an S phase arrest (Fig. 1D and F). Interestingly, whereas exhibiting cytoplasmic localizations in untreated cells, cisplatin treatment resulted in translocation of all Apaf-1 forms to the nucleus (Fig. 1D).
Figure 1.

Apaf-1 main isoforms translocate to the nucleus and mediate cell-cycle arrest upon genotoxic stress. (A) Schematic diagram of the Apaf-1 isoforms examined in this study. The CARD domain, the CED-4 homology domain and the WD-repeats are indicated. (B) Expression of indicated Apaf-1 constructs in transduced apaf-1−/− MEFs as determined by immunoblotting with an anti-Apaf-1 antibody. (C) Cytochrome c-dependent processing of procaspase-9 by the different Apaf-1 isoforms.35S-labeled procaspase-9 was incubated with S100 extracts of apaf-1−/− MEFs cells expressing the indicated constructs for 30 min at 30°C in the presence or absence of 8 μg/ml cytochrome c and 1 mM dATP. The samples were then analyzed by SDS-PAGE and autoradiography. (D) Nuclear translocation of Apaf-1 isoforms upon DNA damage. WT MEFs or apaf-1−/− MEFs expressing the indicated constructs, were treated or not with cisplatin (20 μM, 24 h), fixed in Triton X-100 and immunolabeled with anti-Apaf-1 antibody (red). Nuclei were stained with DAPI. Numbers indicate percentage of cells with nuclear Apaf-1 (means ± s.e.m.); n = 3. (E, F) All Apaf-1 isoforms complement defective genotoxic stress-induced Chk1 activating phosphorylation and S phase arrest in apaf-1−/− MEFs. (E) WT MEFs or apaf-1−/− MEFs cells expressing the indicated constructs were treated or not with cisplatin (20 μM, 24 h) and the phosphorylation of Chk1 (serine 317) was assayed by western blotting. (F) WT MEFs or apaf-1−/− MEFs cells expressing the indicated constructs were treated or not with cisplatin (20 μM, 24 h) and the S phase arrest was assessed by cell-cycle analysis.
Upon DNA damage, Chk1, a major effector of intra- S checkpoint signaling,14 is activated through phosphorylation at serine 317 or 345.15 In lane with the study of Kroemer and coworkers,9 cisplatin-treated apaf-1−/− MEFs exhibited a reduced Chk1 phosphorylation compared to WT MEFs (Fig. 1E). This observation confirms that Apaf-1 is involved in the optimal activation of the essential cell cycle checkpoint kinase Chk1. As a consequence, the extent of cisplatin-induced S phase arrest was largely reduced in apaf-1−/− MEFs (Fig. 1F). However, expression of the different Apaf-1 isoforms resulted in a notably increased Chk1 phosphorylation upon cisplatin treatment (Fig. 1E). Accordingly, all the tested Apaf-1 isoforms were able to restore the cisplatin-induced cell cycle arrest in S phase of the apaf-1−/− MEFs (Fig. 1F). Combined, our results confirm the implication of Apaf-1 in DNA damage‑induced cell cycle arrest and indicate that the main Apaf-1 isoforms, possibly as a result of their translocation to the nucleus, can mediate Chk1 activation, actively effecting cell cycle arrest at the S phase following genotoxic stress.
Apaf-1 associates with Nup107
The mechanisms that trigger Apaf-1 nuclear translocation upon DNA damage are unknown. To determine whether p53 regulates the genotoxic stress-dependent Apaf-1 nuclear translocation, we studied the effect of cisplatin treatment on Apaf-1 cellular localization in A549 (p53+/+) and H1299 (p53−/−) cells. As shown in Figure 2A, cisplatin induced translocation of Apaf-1 from the cytoplasm to the nucleus in both A549 and H1299 cells, indicating that it constitutes a p53 independent mechanism.
Figure 2.
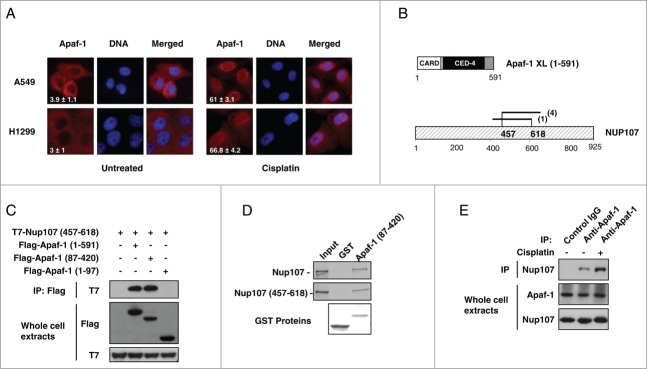
Apaf-1 associates with the nucleoporin Nup107. (A) Apaf-1 nuclear translocation upon genotoxic stress is independent of p53. A549 (p53+/+) and H1299 (p53−/−) cells were treated or not with cisplatin (20 μM, 24 h) and immunolabeled with anti-Apaf-1 antibody (red). Nuclei were stained with DAPI. Numbers indicate percentage of cells with nuclear Apaf-1 (means ± s.e.m.); n = 3. (B) Top: Schematic representation of Apaf-1 XL (1-591), which was used as bait for the yeast 2-hybrid screening. Bottom: Overlapping fragments of the nucleoporin Nup107 (NM_020401) that interact with Apaf-1 in the yeast-2-hybrid assay. Black lines indicate the fragments of Nup107 that interact with Apaf-1. The number of times the fragments were identified in the screen are indicated. The minimal interacting domain is from aa 457 to aa 618. (C) Nup107 (457–618) interacts with the CED-4 domain of Apaf-1. 293T cells were cotransfected with T7-Nup107 (457–618) together with the indicated Apaf-1 Flag-tagged constructs. Cells were lysed and the lysates were subjected to anti-Flag immunoprecipitation (IP) followed by immunoblotting (IB) with anti-T7. The expression of different T7- or Flag-tagged constructs was determined by direct immunoblotting. (D) In vitro interaction of Apaf-1 with Nup107. 35S-labeled Nup107 or Nup107 (457-618) (lane 1, 10% input) were incubated with an equal amount of glutathione Sepharose beads bound to an equal amount of GST (lane 2) or GST- Apaf-1 (87-420) (lane 3). Bound proteins were eluted and analyzed by SDS-PAGE followed by autoradiography. (E) Endogenous Apaf-1 interacts with endogenous Nup107. Whole cell extracts derived from A549 cells treated or not with cisplatin were immunoprecipitated with a control antibody or anti-Apaf-1 antibody and analyzed by immunoblotting with anti-Nup107 antibody.
To gain access into the Apaf-1 signaling pathway, we performed a yeast 2-hybrid assay using the WD40-deleted Apaf-1XL (residues 1–591) as bait to screen a highly complex, random primed cDNA library from CEMC7 cells. By this approach, several overlapping fragments of the nucleoporin Nup107 (GenBankTM accession number NM_020401) were identified, allowing us to narrow down the precise interaction domain of Nup107 to amino acids 457–618 (Fig. 2B). To confirm whether the interaction we detected by yeast 2-hybrid analysis could take place in mammalian cells, we transfected 293T cells with expression vectors encoding T7-tagged Nup107 (457-618) and Flag-tagged Apaf-1 (1–591). As shown in Figure 2C, Nup107 (457-618) was detectable in Flag immunoprecipitates. To extend the characterization of the Nup107 (457-618)-Apaf-1 interaction, we expressed T7-tagged Nup107 (457-618) together with several Flag-tagged truncations of Apaf-1. As shown in Figure 2C, a deletion mutant containing only the CED-4 homologous domain of Apaf-1 (residues 87–420) was still able to co-precipitate with Nup107 (457–618).
To rule out the possibility that other proteins were necessary for the detected interaction, we analyzed the ability of a purified GST-Apaf-1 (87–420) fusion protein to associate with in vitro translated 35S-labeled Nup107 or Nup107 (457–618). In agreement with a direct interaction, 35S-labeled Nup107 or Nup107 (457–618) bound to the GST-Apaf-1 (87–420) fusion protein but not the GST control (Fig. 2D).
To confirm the Apaf-1-nup107 interaction at the endogenous level, endogenous Apaf-1 was immunoprecipitated from A549 whole cell extracts using a control or anti-Apaf-1 antibody. As shown in Figure 2E, Nup107 was co-immunoprecipitated with Apaf-1, but not with a control antibody, indicating that endogenous Apaf-1 and Nup107 interact physiologically. Interestingly, increased association of Apaf-1 and Nup107 was detected after a 24-hour treatment with cisplatin (Fig. 2E). This elevated interaction was not due to upregulation of Apaf-1 or Nup107 proteins, suggesting post-translational modification event(s). Therefore, our data indicate that Apaf-1 interacts through its CED-4 domain with the central domain of NUP107, and this interaction is favored upon genotoxic stress.
ATR-dependent phosphorylation of Apaf-1 regulates its interaction with Nup107
Phosphorylations of apoptosome-associated proteins have been described, that are known to regulate cytochrome c-induced caspase activation5,16 and Apaf-1 is known to be phosphorylated both in vivo and in vitro,17,18 leading us to examine whether Apaf-1 could be phosphorylated upon genotoxic stress. For that purpose, extracts from A549 cells treated or not with cisplatin were immunoprecipitated using an anti-Apaf-1 antibody followed by immunoblotting with an anti-phosphoserine or anti-phosphothreonine antibody. As shown in Figure 3A, significant level of serine phosphorylated Apaf-1 was detected in unstimulated cells. Interestingly, this level increased following treatment with cisplatin (Fig. 3A). However, Apaf-1 only appeared to be weakly phosphorylated at threonine residue(s) prior or following cisplatin treatment (Fig. 3A). Since cisplatin treatment increases Apaf-1-Nup107 binding (Fig. 2D), this observation suggests that phosphorylation of Apaf-1 at serine residue(s) could promote its interaction with Nup107 and therefore its nuclear translocation following genotoxic stress. To test this hypothesis, we used the pan-kinase inhibitor staurosporine to examine whether protein kinase inhibition could interfere with genotoxic stress-induced Apaf-1 nuclear translocation. For that purpose, we studied Apaf-1 location in the retinoblastoma (pRb) deficient tumorigenic breast cancer cell line MDA-MB436. We use this cell line as, in contrast to pRb+ cells, pRb− cells are refractory to the G1-arresting abilities of staurosporine.19 As shown in Figure 3B, treatment of the MDA-MB436 cells with cisplatin resulted in nuclear relocalization of Apaf-1, indicating that the retinoblastoma pathway is not involved in Apaf-1 nuclear translocation. Interestingly, treatment with 4 nM of staurosporine abolished cisplatin-induced Apaf-1 nuclear accumulation (Fig. 3B). Moreover, staurosporine treatment prevented Apaf-1 phosphorylation upon genotoxic stress (Fig. 3C) and drastically reduced its interaction with Nup107 (Fig. 3D). Combined, these data indicate that Apaf-1 phosphorylation is a prerequisite for its nuclear translocation.
Figure 3 (See previous page).

ATR-dependent Apaf-1 phosphorylation increases its association with Nup107. (A) Apaf-1 is phosphorylated at serine residue(s) following genotoxic stress. Whole cell extracts derived from A549 cells treated or not with cisplatin were immunoprecipitated with an anti-Apaf-1 antibody and analyzed by immunoblotting with anti-phosphoserine, anti-phosphothreonine or anti-Apaf-1 (as an immunoprecipitation control) antibody. (B) The kinase inhibitor staurosporine prevents Apaf-1 nuclear translocation upon genotoxic stress. MDA-MB436 cells were treated or not with cisplatin (20 μM, 24 h) in the presence or absence of staurosporine (4 nM). After fixation, cells were immunolabeled with anti-Apaf-1 antibody (red). Nuclei were stained with DAPI. Numbers indicate percentage of cells with nuclear Apaf-1 (means ± s.e.m.); n = 3. (C) Staurosporine prevents Apaf-1 phosphorylation upon genotoxic stress. Lysates of cells treated as in (B) were immunoprecipitated with anti-Apaf-1 antibody and analyzed by immunoblotting with anti-phosphoserine or anti-Apaf-1 (as an immunoprecipitation control) antibody. (D) Staurosporine reduces Apaf-1-Nup107 interaction. Lysates of cells treated as in (B) were immunoprecipitated with anti-Apaf-1 antibody and analyzed by immunoblotting with anti-Nup107 antibody. (E) Inhibition of ATR drastically reduces Apaf-1 nuclear translocation upon cisplatin treatment. A549 cells were left untreated or were pretreated for 1 h with DMSO, 10 μM NU-7026, 10 μM KU-55933, 10 μM VE-821, 20 μM U0126, 20 μM SP600125, 50 μM LY294002 or 10 μM BI-D1870. Cells were then treated or not with cisplatin (20 μM, 24 h), fixed and nuclear Apaf-1 assessed by immunofluorescence (means ± s.e.m., n = 3). (F) Inhibition of ATR prevents Apaf-1 phosphorylation upon genotoxic stress. A549 cells pretreated for 1 h with vehicle or 10 μM VE-821 were treated or not with cisplatin (20 μM, 24 h), lysed and the lysates immunoprecipitated and analyzed as in (C). (G) Inhibition of ATR reduces Apaf-1-Nup107 interaction. Lysates of cells treated as in (F) were immunoprecipitated with anti-Apaf-1 antibody and analyzed as in (D). (H) Deregulation of ATR prevents Apaf-1 phosphorylation upon genotoxic stress. A549 cells were transfected with mock or ATR specific siRNAs. After 48 h, cells were treated or not with cisplatin (20 μM, 24 h), lysed and the lysates immunoprecipitated and analyzed as in (C). (I) Deregulation of ATR reduces Apaf-1-Nup107 interaction. A549 cells were transfected with mock or ATR specific siRNAs. After 48 h, cells were treated or not with cisplatin (20 μM, 24 h), lysed and the lysates immunoprecipitated with anti-Apaf-1 antibody and analyzed as in (D). (J) Deregulation of ATR prevents Apaf-1 nuclear translocation upon genotoxic stress. A549 cells were transfected with mock or ATR specific siRNAs. After 48 h, cells were treated or not with cisplatin (20 μM, 24 h). After fixation, cells were immunolabeled with anti-Apaf-1 antibody (red). Nuclei were stained with DAPI. Numbers indicate percentage of cells with nuclear Apaf-1 (means ± s.e.m.); n = 3.
Genotoxic stress in response to cisplatin activates numerous signal transduction pathways. Indeed, beside the apical PI3 kinase related kinases (PIKKs) ATM, ATR, and DNA-PK, cisplatin is known to activate several effectors, such as the MAPK (mitogen-activated protein kinase) pathway members extracellular signal-regulated kinase (ERK) and c-Jun-N-terminal kinase 1 (JNK1).20 To determine the identity of the kinases(s) involved in DNA damage-induced Apaf-1 nuclear translocation, we screened a panel of kinase inhibitors to assess their effects on Apaf-1 translocation upon cisplatin treatment. Interestingly, only the ATR-selective inhibitor VE-821 was able to prevent cisplatin-induced Apaf-1 nuclear accumulation (Fig. 3E). Inhibition of ATR prevented Apaf-1 serine phosphorylation and markedly reduced its association with Nup107 following cisplatin treatment (Fig. 3F and G), whereas the other kinase inhibitors failed to do so (not shown). We then further assessed the role of ATR in Apaf-1 phosphorylation-dependent nuclear relocalization upon cisplatin treatment using RNA interference experiments. As shown in Figure 3H, silencing of ATR drastically reduced cisplatin-induced phosphorylation of Apaf-1 as well as its interaction with Nup107 (Fig. 3I). Consequently, as previously observed,9 nuclear accumulation of Apaf-1 was reduced in ATR-depleted cells following cisplatin treatment (Fig. 3J). However, using purified components, we failed to demonstrate a direct role for ATR in phosphorylating Apaf-1 (not shown), suggesting that adaptor/scaffold molecules or other kinase(s) downstream of ATR might be required. Still, taken together, our data indicate that ATR regulates the phosphorylation-dependent Apaf-1 nuclear translocation upon cisplatin treatment.
Nuclear import of Apaf-1 involves binding with Nup107
We next sought to determine if the interaction with Nup107 contributes to nuclear import of Apaf-1 following genotoxic stress. For that purpose, we first explored the functional consequence of Nup107 depletion on the cisplatin-induced distribution of Apaf-1 in A549 cells. Small interfering RNA (siRNA) directed against Nup107, but not mock siRNA, effectively depleted Nup107 whereas the level of Apaf-1 did not change (Fig. 4A and not shown). As previously shown,21 immunofluorescence analysis of mock-transfected A549 cells with α-Nup107 demonstrated nuclear rim staining (Fig. 4B). In agreement with the data shown in Figure 4A, the nuclear rim staining was no longer detected in the Nup107 siRNAs-transfected cells (Fig. 4B). Interestingly, whereas Apaf-1 nuclear translocation occurred in mock siRNA transfected cells, Apaf-1 did not efficiently translocate to the nucleus in Nup107-knockdown cells following cisplatin treatment, and remained essentially cytoplasmic (Fig. 4B). These data point to a crucial role for Nup107 in DNA damage-elicited nuclear translocation of Apaf-1.
Figure 4.

Deregulation of Nup107 prevents Apaf-1 role in DNA damage checkpoint. (A) A549 cells transfected with 2 rounds of control or Nup107 siRNAs were harvested and lysed 48 h after second round of siRNA transfection. Cell lysates were analyzed by immunoblotting with anti-Nup107 or anti-β-actin antibody. (B) Silencing of Nup107 prevents Apaf-1 nuclear translocation upon genotoxic stress. A549 cells transfected with 2 rounds of control siRNA or Nup107 siRNAs were treated or not with cisplatin (20 μM, 24 h) 24 h after second round of siRNA transfection. After fixation, cells were immunolabeled with anti-Apaf-1 antibody (red) or anti-Nup107 antibody (green). Nuclei were stained with DAPI. Numbers indicate percentage of cells with nuclear Apaf-1 (means ± s.e.m.); n = 3. (C) Expression of Nup107 (457–618) prevents Apaf-1 nuclear translocation upon genotoxic stress. A549 cells expressing Flag-Nup107 (457-618) were treated or not with cisplatin (20 μM, 24 h), fixed in Triton X-100 and immunolabeled with anti-Apaf-1 antibody (red) or anti-Flag antibody (green). Nuclei were stained with DAPI. Numbers indicate percentage of cells with nuclear Apaf-1 (means ± s.e.m.); n = 3. (D, E) Expression of Nup107 (457-618) prevents S phase arrest and Chk1 activating phosphorylation upon genotoxic stress. (D) A549 cells expressing Flag-Nup107 (457–618) were treated or not with cisplatin (20 μM, 24 h) and the cell-cycle distribution of viable cells was assessed by cell-cycle analysis. (E) A549 cells expressing Flag-Nup107 (457–618) were treated or not with cisplatin (20 μM, 24 h) and the phosphorylation of Chk1 (serine 317) was assayed by protein gel blotting.
If the Nup107-Apaf-1 interaction is required for genotoxic stress-induced Apaf-1 nuclear accumulation, one can expect that preventing this interaction would prevent Apaf-1 nuclear import following DNA damage. To address this hypothesis, we expressed Nup107 (457–618) as a “decoy” to interfere with the interaction between endogenous Apaf-1 and Nup107. Immunofluorescence microscopy of Nup107 (457–618) transfected cells indicated that the construct was largely cytoplasmic. (Fig. 4C) Interestingly, Nup107 (457–618) exhibited a cellular localization similar to that of endogenous Apaf-1. As shown in Figure 4C, Apaf-1 failed to accumulate in the nucleus following cisplatin treatment when Nup107 (457–618) was expressed, and both proteins exhibited fluorescence overlap, suggesting that Nup107 (457-618) “trapped” Apaf-1 in the cytoplasm. Fitting with the hypothesis that Apaf-1 nuclear accumulation, upon genotoxic stress, is required for its cell cycle function, Nup107 (457–618) expressing cells failed to enter S phase after cisplatin treatment (Fig. 4D). This absence of cell cycle arrest correlated with an absence of activating phosphorylation of Chk1 (Fig. 4E), indicating that Chk1 acts downstream of Apaf-1 nuclear accumulation. Together, these data suggest that Apaf-1 is imported in the nucleus by interacting with Nup107 and this nuclear accumulation is essential for Apaf-1 role in the DNA damage checkpoint.
Discussion
Apaf-1, an adaptor protein critically involved in mitochondrial cell death, is known to possess an essential role in mediating the S phase arrest following DNA damage.9 Whereas this function seems to require Apaf-1 nuclear accumulation, the mechanisms responsible for Apaf-1 nuclear import upon genotoxic stress, which is devoid of a putative nuclear localization signal (NLS), are unknown. Different transcripts of apaf-1 are produced by alternative splicing, containing or not NH2- or COOH-terminal inserts, but the respective role of the different Apaf-1 isoforms in cell cycle regulation remains to be clarified. In this report, we show that the main expressed forms of Apaf-1 can all accumulate in the nucleus and mediate cell cycle arrest following DNA damage. The cell cycle arrest induced by the Apaf-1 isoforms seemed to be dependent on Chk1 activation, as all the Apaf-1 forms were able to complement Chk1 phosphorylating activation in apaf-1−/− MEFs. Previous studies indicate that only the isoforms containing an additional WD40 are able to interact with cytochrome c and activate caspase-9, whereas the 11-amino acid insert between the CARD and the CED-4 domain was not required for binding to cytochrome c or caspase-9 activation.6 Therefore, contrary to what observed for Apaf-1 apoptotic function, our data exclude a role for Apaf-1 NH2- or COOH-terminal inserts in nuclear translocation and S phase arrest upon genotoxic stress.
Using a yeast 2-hybrid approach, we identified the nucleoporin Nup107 as a binding partner of Apaf-1. Nup107 is part of a conserved nuclear pore sub-complex that is crucial for nuclear pore complex (NPC) assembly.22,23 The NPC is a large, multiprotein assembly located at the fusion points between the outer and the inner nuclear membranes that mediates macromolecular traffic between the nucleus and the cytoplasm.24 The Nup107 complex, which in vertebrates is formed by 9 subunits, is now known to be involved in a wide array of functions, including macro-molecular transport, transcription, and DNA repair (see25 for review). Here we show that Apaf-1 binds, through its CED-4 domain, to the central domain of Nup107, and that this interaction is responsible for Apaf-1 nuclear accumulation upon genotoxic stress. In lane with this hypothesis, expression of the central domain of Nup107, Nup107 (457-618), prevented DNA damage-induced Apaf-1 nuclear translocation, indicating that binding of Apaf-1 to the central domain of endogenous Nup107 is a prerequisite for its nuclear import. Moreover, because expression of Nup107 (457-618) prevents both DNA damage-induced activation of Chk1 and S phase arrest, our data confirm that nuclear translocation of Apaf-1 is required for its cell cycle function. Increasing amount of data indicate that the NPC contributes to DNA repair. Indeed, members of the Nup107 complex have been demonstrated to potentiate the double strand breaks repair processes by inducing the recruitment of damaged DNA and sumoylation enzymes to nuclear pores.26,27 Thus, our data confirm the crucial role of the Nup107 complex in the DNA damage response.
Although Nup107 binds to the CED-4 domain of Apaf-1, we do not know if this binding involves a domain essential for Apaf-1 oligomerization, like the binding pocket for dATP, which is crucial for apoptosome assembly. The discovery of Apaf-1 dual implication in caspase activation and cell cycle blockade would call for the identification of approaches that facilitate the dissection of Apaf-1-dependent pathways. Indeed, at this moment, very little is known about the pattern of crosstalk between these 2 signaling pathways. Interestingly, QM31, a recently developed small molecule that binds to Apaf-1, has been shown to inhibit these 2 functions of Apaf-1.28 Even known this remains to be demonstrated, it is possible that this inhibitor could alter Apaf-1 structure in a way that it would prevent Apaf-1 interaction with some of its molecular partners. Therefore, this novel class of inhibitor provides a new molecular tool as lead to identify compounds that could be used as probes to study Apaf-1 interaction with putative or identified interactors and understand their respective roles in Apaf-1 signaling mechanisms.
Our study indicates that Apaf-1 appears to interact with Nup107 in a phosphorylation-dependent manner that is regulated by ATR. Indeed, Apaf-1 phosphorylation levels increased upon genotoxic stress, and blockade of cisplatin-induced Apaf-1 phosphorylation by the ATR-specific inhibitor VE-821 or deregulation of ATR compromised its interaction with Nup107 and its nuclear translocation. Thus, one might speculate that phosphorylation of Apaf-1 can result in conformational changes that favor its interaction with Nup107. Our data confirm the crucial role of ATR, through nuclear translocation of Apaf-1, in the proper execution of the DNA damaging response in terms of S phase checkpoint arrest. Biochemically, however, we were not able to demonstrate a direct phosphorylation of Apaf-1 by ATR. This suggests that adaptor or scaffold proteins may modulate ATR activity in phosphorylating Apaf-1, or that additional kinase(s) that function in downstream signaling are required. Apaf-1 is known to be phosphorylated at S256 by the 90-kDA ribosomal S6 kinase (Rsk).17 However, inhibition of Rsk did not prevent Apaf-1 nuclear accumulation upon cisplatin treatment (this work) and a S256A Apaf-1 mutant was still able to interact with Nup107 and accumulate in the nucleus following genotoxic stress (data not shown). Therefore, the precise site(s) of phosphorylation remain to be identified. The Nup107 complex is also known to be phosphorylated in a cell-cycle-dependent manner.29 However, we could not detect an increase of the phosphorylation state of Nup107 following genotoxic stress (data not shown). Even known this represents principally a mitotic event, it is therefore possible that phosphorylation state of the Nup107 complex can also regulate its interaction with Apaf-1.
Proteins containing nonclassical NLS are known to be transported through the nuclear pore in an importin β-dependent fashion.30 However, we haven't been able to detect any interaction between Apaf-1 and importin α or β (Jagot-Lacoussiere L. and Poyet J.L., personal communication). Therefore, Apaf-1 could, as in the case of β-catenin,31 translocate to the nucleus through direct binding of nucleoporins.
In addition to its cell-cycle function, Apaf-1 has recently been demonstrated to participate to the centrosomal microtubule nucleation process, and spindle assembly.8 Interestingly, the Nup107 complex is known to relocalize to spindle fibers during prophase, where it affects spindle assembly, kinetochore function, and the spindle assembly checkpoint.32 Therefore, our finding that Apaf-1 interacts with Nup107 may have potentially important implications for understanding the molecular basis for the abnormal bipolar mitotic spindle formation observed in Apaf-1 deficient cells.8
In summary, our results show that genotoxic stress-induced Apaf-1 nuclear translocation, which occurs in a p53- and pRb-independent mechanism, is mediated through direct interaction with the nucleoporin Nup107, and that this relocalization is crucial for the DNA damage checkpoint role of Apaf-1. Although future studies will be needed to provide the molecular mechanisms involved in this cell-cycle function, the present study confirms a crucial role for the Nup107 complex in the DNA damage response, indicating that nucleoporins, by modulating nuclear trafficking of signal molecules, contribute significantly to the intra‑S‑phase checkpoint, and therefore to preservation of genomic stability.
Material and Methods
Cell culture
Cells were cultivated either in Dulbecco's modified Eagle medium (MEFs, A549, MDA-MB436 or 293 T cells) or RPMI 1640 (H1299 cells) (Gibco), supplemented with 10% fetal bovine serum, 200 μg/ml penicillin and 100 μg/ml streptomycin sulfate.
Materials and antibodies
Protease, kinases and phosphatase inhibitors, RNAse A, staurosporine and cisplatin were purchased from Sigma, Santa Cruz and APIs Chemical. Antibodies used for immunoprecipitation and Western blotting were: mouse anti-Apaf-1 (Abcam), rabbit anti-Apaf-1 (RD Systems), mouse anti-Flag M2 (sigma), mouse anti-T7 (Sigma), mouse anti-T7 HRP-conjugated (Sigma), mouse anti-phosphoserine (Invitrogen), mouse anti-phosphothreonine (Invitrogen), mouse anti-β-tubulin (BD PharMingen). Rabbit anti-Nup107 was a gift from Dr. Valerie Doye.
Plasmid constructs
Apaf-1 constructs encoding Myc-tagged Apaf-1XL, LN, LC and S were a gift from Prof. Gabriel Nùñez. The complete cDNA of Nup107 was isolated from a human HeLa cDNA library by the polymerase chain reaction (PCR) using Nup107 specific primers. Truncated mutants of Apaf-1 or Nup107 were generated by PCR using modified complementary PCR adapter primers. Flag- and T7-epitope tagging was done by cloning the PCR generated cDNAs of the respective genes in-frame into pcDNA3-Flag and pcDNA3-T7 vectors (Invitrogen), respectively. GST-tagging was done by cloning the PCR generated cDNAs together with GST cDNA in-frame into pGEX-4T (Amersham).
Lentiviral transduction
Apaf-1XL, LC, LN and S cDNAs were cloned into the pLVX-IRES-ZsGreen lentiviral vector (Clontech). To produce lentiviral particles, 293T cells were transfected with the pLVX-IRES-ZsGreen lentiviral constructs together with the packaging plasmids psPAX2 and pMD2G (Addgene) using FuGENE HD Transfection Reagent (Promega). Lentiviral supernatants were harvested 48 h after transfection, filtered before being used to infect apaf−/− MEFs with 4 μg/ml of polybrene. 72 h after transduction, cells were diluted for immunoblot selection of individual clones. Clones with similar Apaf-1 levels to WT MEFs were selected, expanded, and analyzed.
Yeast two-hybrid screening procedure
Two-hybrid screen was performed using a cell-to-cell mating as described earlier33 using WD40-deleted Apaf-1 (residues 1–591) as bait and a cDNA library from T-cell leukemia cells CEMC7 poly(A+) RNA. Positive clones were obtained using the HIS3 reporter gene reporter gene selection.
In vitro caspase-9 assay
Cytosolic extracts were prepared by resuspending the cells in buffer A (20 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1.5 mM, MgCl2, 1 mM sodium EDTA, 1 mM sodium EGTA, 1 mM dithiothreitol). Cells were lysed by syringing (×20) using a 21-gauge needle followed by centrifugation at 17 000 g for 30 min at 4°C. Labeled procaspase-9 was prepared by in vitro transcription and translation in the presence of [35S]methionine using a TNT Quick System of coupled transcription-translation (Promega). Ten μg of the cytosolic extracts were incubated with 1 μl of in vitro translated procaspase-9 in the presence or without dATP (1 mM) and cytochrome c (8 μg/ml) in a final volume of 25 μl for 30 min at 30°C. The reactions were then boiled in SDS sample buffer and subjected to SDS-PAGE analysis followed by autoradiography.
Cell-cycle analysis
5 × 105 cells were washed twice with ice-cold PBS, fixed in cold 70% ethanol for at least 1 hour at 4°C, washed twice in cold PBS, treated with 200 U per mL DNAse-free RNase A for 30 minutes at 37°C, and stained with 25 μg/mL PI. Distribution of cell-cycle phases was determined within the hour using PI fluorescence emission (15 000 events) on FACScalibur and analyzed with Modfit Software (BD Biosciences).
Immunocytochemistry
Cells, grown on coverslips, were washed twice with PBS, fixed in 4% PFA for 10 min and permeabilized with 0.2% Triton X-100/PBS for 5 min. After blocking with 2% BSA/PBS for 1 h, the cells were incubated with specific antibodies diluted in 1% BSA/PBS for 1 h. Cells were washed 3 times in blocking buffer and incubated for 1 h with labeled anti-mouse or anti-rabbit secondary antibodies (Alexa Fluor 488 or Alexa Fluor 568; Molecular Probe). After rinsing in blocking buffer and PBS, nuclei were counterstained with DAPI. Images were acquired using a Zeiss Axiovert 200 M microscope or by confocal microscopy on a Zeiss LSM 510 META confocal laser microscope (Zeiss, Oberkochen, Germany) with a Plan Apochromat 63 × N.A.1.4 oil-immersion objective using the LSM510 software v4.0 (Zeiss).
Transfection, immunoprecipitation and immunoblot analysis and GST pull-down analysis
293 T cells (5 × 105 cells) in 60 mm dishes were transiently transfected with the expression plasmids using the JetPei (Polyplus Transfection) method as per the manufacturer's instructions. Cells were lysed, and the cellular lysates were immunoprecipitated and Western blotted as described before.34 GST pull-down were performed as previously described.35 For siRNA knockdown, cells were transfected using HiPerFect Transfection Reagent (Qiagen) according to the manufacturer’ instructions. After 24 h, another round of siRNA transfections were carried out, and after 48 h, cells were processed for further analysis. siRNA oligo sequences used were as follows: ATR, 5′-AACCUCCGUGAUGUUGCUUGA-3′ Nup107, 5′-CUGCGAAUACACUUUCCUCTT-3′ and scrambled, 5′-UAGAUACCAUGCACAAUCCTT-3′.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr. Valerie Doye for providing anti-Nup107 antibody and Prof. Gabriel Nùñez for Apaf-1 cDNAs constructs.
References
- 1. Cohen GM. Caspases: the executioners of apoptosis. Biochem J 1997; 326 (Pt 1):1-16; PMID:9337844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Song Z, Steller H. Death by design: mechanism and control of apoptosis. Trends Cell Biol 1999; 9:M49-52; PMID:10611682; http://dx.doi.org/ 10.1016/S0962-8924(99)01670-0 [DOI] [PubMed] [Google Scholar]
- 3. Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997; 90:405-13; PMID:9267021; http://dx.doi.org/ 10.1016/S0092-8674(00)80501-2 [DOI] [PubMed] [Google Scholar]
- 4. Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol 2008; 9:532-42; PMID:18568041; http://dx.doi.org/ 10.1038/nrm2434 [DOI] [PubMed] [Google Scholar]
- 5. Schafer ZT, Kornbluth S. The apoptosome: physiological, developmental, and pathological modes of regulation. Dev Cell 2006; 10:549-61; PMID:16678772; http://dx.doi.org/ 10.1016/j.devcel.2006.04.008 [DOI] [PubMed] [Google Scholar]
- 6. Benedict MA, Hu Y, Inohara N, Nunez G. Expression and functional analysis of Apaf-1 isoforms. Extra Wd-40 repeat is required for cytochrome c binding and regulated activation of procaspase-9. J Biol Chem 2000; 275:8461-8; PMID:10722681; http://dx.doi.org/ 10.1074/jbc.275.12.8461 [DOI] [PubMed] [Google Scholar]
- 7. Hu Y, Benedict MA, Ding L, Nunez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J 1999; 18:3586-95; PMID:10393175; http://dx.doi.org/ 10.1093/emboj/18.13.3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ferraro E, Pesaresi MG, De Zio D, Cencioni MT, Gortat A, Cozzolino M, Berghella L, Salvatore AM, Oettinghaus B, Scorrano L, et al. . Apaf1 plays a pro-survival role by regulating centrosome morphology and function. J Cell Sci 2011; 124:3450-63; PMID:21984814; http://dx.doi.org/ 10.1242/jcs.086298 [DOI] [PubMed] [Google Scholar]
- 9. Zermati Y, Mouhamad S, Stergiou L, Besse B, Galluzzi L, Boehrer S, Pauleau AL, Rosselli F, D'Amelio M, Amendola R, et al. . Nonapoptotic role for Apaf-1 in the DNA damage checkpoint. Mol Cell 2007; 28:624-37; PMID:18042457; http://dx.doi.org/ 10.1016/j.molcel.2007.09.030 [DOI] [PubMed] [Google Scholar]
- 10. Hausmann G, O'Reilly LA, van Driel R, Beaumont JG, Strasser A, Adams JM, Huang DC. Pro-apoptotic apoptosis protease-activating factor 1 (Apaf-1) has a cytoplasmic localization distinct from Bcl-2 or Bcl-x(L). J Cell Biol 2000; 149:623-34; PMID:10791976; http://dx.doi.org/ 10.1083/jcb.149.3.623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruiz-Vela A, Gonzalez de Buitrago G, Martinez AC. Nuclear Apaf-1 and cytochrome c redistribution following stress-induced apoptosis. FEBS Lett 2002; 517:133-8; PMID:12062423; http://dx.doi.org/ 10.1016/S0014-5793(02)02607-8 [DOI] [PubMed] [Google Scholar]
- 12. Sakai T, Liu L, Teng X, Mukai-Sakai R, Shimada H, Kaji R, Mitani T, Matsumoto M, Toida K, Ishimura K, et al. . Nucling recruits Apaf-1/pro-caspase-9 complex for the induction of stress-induced apoptosis. J Biol Chem 2004; 279:41131-40; PMID:15271982; http://dx.doi.org/ 10.1074/jbc.M402902200 [DOI] [PubMed] [Google Scholar]
- 13. Besse B, Cande C, Spano JP, Martin A, Khayat D, Le Chevalier T, Tursz T, Sabatier L, Soria JC, Kroemer G. Nuclear localization of apoptosis protease activating factor-1 predicts survival after tumor resection in early-stage non-small cell lung cancer. Clin Cancer Res 2004; 10:5665-9; PMID:15355891; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-0415 [DOI] [PubMed] [Google Scholar]
- 14. Robinson HM, Jones R, Walker M, Zachos G, Brown R, Cassidy J, Gillespie DA. Chk1-dependent slowing of S-phase progression protects DT40 B-lymphoma cells against killing by the nucleoside analogue 5-fluorouracil. Oncogene 2006; 25:5359-69; PMID:16619043; http://dx.doi.org/ 10.1038/sj.onc.1209532 [DOI] [PubMed] [Google Scholar]
- 15. Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 2001; 21:4129-39; PMID:11390642; http://dx.doi.org/ 10.1128/MCB.21.13.4129-4139.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kurokawa M, Kornbluth S. Caspases and kinases in a death grip. Cell 2009; 138:838-54; PMID:19737514; http://dx.doi.org/ 10.1016/j.cell.2009.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim J, Parrish AB, Kurokawa M, Matsuura K, Freel CD, Andersen JL, Johnson CE, Kornbluth S. Rsk-mediated phosphorylation and 14-3-3varepsilon binding of Apaf-1 suppresses cytochrome c-induced apoptosis. EMBO J 2012; 31:1279-92; PMID:22246185; http://dx.doi.org/ 10.1038/emboj.2011.491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martin MC, Allan LA, Lickrish M, Sampson C, Morrice N, Clarke PR. Protein kinase A regulates caspase-9 activation by Apaf-1 downstream of cytochrome c. J Biol Chem 2005; 280:15449-55; PMID:15703181; http://dx.doi.org/ 10.1074/jbc.M414325200 [DOI] [PubMed] [Google Scholar]
- 19. McGahren-Murray M, Terry NH, Keyomarsi K. The differential staurosporine-mediated G1 arrest in normal versus tumor cells is dependent on the retinoblastoma protein. Cancer Res 2006; 66:9744-53; PMID:17018634; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-1809 [DOI] [PubMed] [Google Scholar]
- 20. Vyas D, Laput G, Vyas AK. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. OncoTargets Ther 2014; 7:1015-23; PMID:24959088; http://dx.doi.org/ 10.2147/OTT.S60114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Radu A, Blobel G, Wozniak RW. Nup107 is a novel nuclear pore complex protein that contains a leucine zipper. J Biol Chem 1994; 269:17600-5; PMID:8021268 [PubMed] [Google Scholar]
- 22. Boehmer T, Enninga J, Dales S, Blobel G, Zhong H. Depletion of a single nucleoporin, Nup107, prevents the assembly of a subset of nucleoporins into the nuclear pore complex. Proc Natl Acad Sci U S A 2003; 100:981-5; PMID:12552102; http://dx.doi.org/ 10.1073/pnas.252749899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walther TC, Alves A, Pickersgill H, Loiodice I, Hetzer M, Galy V, Hulsmann BB, Kocher T, Wilm M, Allen T, et al. . The conserved Nup107-160 complex is critical for nuclear pore complex assembly. Cell 2003; 113:195-206; PMID:12705868; http://dx.doi.org/ 10.1016/S0092-8674(03)00235-6 [DOI] [PubMed] [Google Scholar]
- 24. Tran EJ, Wente SR. Dynamic nuclear pore complexes: life on the edge. Cell 2006; 125:1041-53; PMID:16777596; http://dx.doi.org/ 10.1016/j.cell.2006.05.027 [DOI] [PubMed] [Google Scholar]
- 25. Gonzalez-Aguilera C, Askjaer P. Dissecting the NUP107 complex: multiple components and even more functions. Nucleus 2012; 3:340-8; PMID:22713280; http://dx.doi.org/ 10.4161/nucl.21135 [DOI] [PubMed] [Google Scholar]
- 26. Nagai S, Dubrana K, Tsai-Pflugfelder M, Davidson MB, Roberts TM, Brown GW, Varela E, Hediger F, Gasser SM, Krogan NJ. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 2008; 322:597-602; http://dx.doi.org/ 10.1126/science.1162790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palancade B, Liu X, Garcia-Rubio M, Aguilera A, Zhao X, Doye V. Nucleoporins prevent DNA damage accumulation by modulating Ulp1-dependent sumoylation processes. Mol Biol Cell 2007; 18:2912-23; PMID:17538013; http://dx.doi.org/ 10.1091/mbc.E07-02-0123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mondragon L, Galluzzi L, Mouhamad S, Orzaez M, Vicencio JM, Vitale I, Moure A, Messeguer A, Perez-Paya E, Kroemer G. A chemical inhibitor of Apaf-1 exerts mitochondrioprotective functions and interferes with the intra-S-phase DNA damage checkpoint. Apoptosis 2009; 14:182-90; PMID:19152031; http://dx.doi.org/ 10.1007/s10495-008-0310-x [DOI] [PubMed] [Google Scholar]
- 29. Glavy JS, Krutchinsky AN, Cristea IM, Berke IC, Boehmer T, Blobel G, Chait BT. Cell-cycle-dependent phosphorylation of the nuclear pore Nup107-160 subcomplex. Proc Natl Acad Sci U S A 2007; 104:3811-6; PMID:17360435; http://dx.doi.org/ 10.1073/pnas.0700058104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kau TR, Way JC, Silver PA. Nuclear transport and cancer: from mechanism to intervention. Nat Rev 2004; 4:106-17; PMID:14732865 [DOI] [PubMed] [Google Scholar]
- 31. Sharma M, Jamieson C, Johnson M, Molloy MP, Henderson BR. Specific armadillo repeat sequences facilitate beta-catenin nuclear transport in live cells via direct binding to nucleoporins Nup62, Nup153, and RanBP2/Nup358. J Biol Chem 2012; 287:819-31; PMID:22110128; http://dx.doi.org/ 10.1074/jbc.M111.299099 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Wozniak R, Burke B, Doye V. Nuclear transport and the mitotic apparatus: an evolving relationship. Cell Mol Life Sci 2010; 67:2215-30; PMID:20372967; http://dx.doi.org/ 10.1007/s00018-010-0325-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fromont-Racine M, Rain JC, Legrain P. Building protein-protein networks by two-hybrid mating strategy. Methods Enzymol 2002; 350:513-24; PMID:12073333; http://dx.doi.org/ 10.1016/S0076-6879(02)50982-4 [DOI] [PubMed] [Google Scholar]
- 34. Rigou P, Piddubnyak V, Faye A, Rain JC, Michel L, Calvo F, Poyet JL. The antiapoptotic protein AAC-11 interacts with and regulates Acinus-mediated DNA fragmentation. EMBO J 2009; 28:1576-88; PMID:19387494; http://dx.doi.org/ 10.1038/emboj.2009.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Piddubnyak V, Rigou P, Michel L, Rain JC, Geneste O, Wolkenstein P, Vidaud D, Hickman JA, Mauviel A, Poyet JL. Positive regulation of apoptosis by HCA66, a new Apaf-1 interacting protein, and its putative role in the physiopathology of NF1 microdeletion syndrome patients. Cell Death Differ 2007; 14:1222-33; PMID:17380155; http://dx.doi.org/ 10.1038/sj.cdd.4402122 [DOI] [PubMed] [Google Scholar]