

Abstract
Background
The majority of tumors trigger macrophage reprogramming from an anti-tumor M1 phenotype towards a pro-tumor M2 phenotype. The M2 phenotype promotes tumor growth. We hypothesized that increasing the number of M1 macrophages in a tumor would limit carcinogenesis and extend the lifespan of the tumor host. The aim of this study was to verify this hypothesis in Ehrlich ascites carcinoma (EAC). The objectives were to evaluate effects of 1) EAC on a macrophage phenotype and NO-producing macrophage activity in vivo; 2) ascitic fluid from mice with EAC on a macrophage phenotype and NO-producing macrophage activity in vitro; and 3) in vitro reprogrammed M1 macrophages on lifespan of mice with EAC.
Material/Methods
The study was conducted using C57BL/6J mice.
Results
Concentration of nitrite, a stable NO metabolite and an index of NO production, was measured spectrophotometrically. Shifts of macrophage phenotype were assessed by changes in NO production as well as by amounts of CD80, a marker of M1 phenotype, and CD206, a marker of M2 phenotype. The CD markers were measured by flow cytometry. Macrophages were reprogrammed towards the M1 phenotype using two reprogramming factors: 0% FBS and 20 ng/ml IFN-γ. The study results showed that 1) EAC inhibited the macrophage NO production in vivo and reprogrammed macrophages towards the M2 phenotype; 2) ascitic fluid of mice with EAC inhibited the macrophage NO production in vitro and reprogrammed macrophages towards the M2 phenotype; and 3) injection of in vitro reprogrammed M1 macrophages into mice with EAC significantly increased the lifespan of mice.
Conclusions
These findings suggest that promising biotechnologies for restriction of tumor growth could be developed based on the in vitro macrophage reprogramming.
MeSH Keywords: Biotechnology; Carcinoma, Ehrlich Tumor; Macrophages
Background
More than 200 types of cancer exist. Each disease is specific but practically all tumors share a common mechanism – pro-tumor transformation of the immune response. As a result of this transformation, immune cells lose their ability to destroy tumor cells and may even begin promoting tumor growth and dissemination [1–3].
Macrophages play a key role in disorders of the immune response in carcinogenesis [1–3]. Macrophages located in the tumor growth area are called tumor-associated macrophages (TAM). Depending on the microenvironment created by the growing tumor and lesion tissue, macrophages acquire a M1 or M2 phenotype [2]. The M1 phenotype of TAM (TAM-M1) exhibits anti-tumor features and contributes to destruction of the tumor by means of nitric oxide (NO) production [4], secretion of proinflammatory cytokines [5–7], activation of natural killer cells [8], and presentation of tumor antigens to lymphocytes [9]. However the majority of tumors, especially malignant ones, trigger mechanisms of macrophage reprogramming. Tumor cells begin producing anti-inflammatory cytokines TGF-β1, IL-4, IL-10, and PGE2 [10–12], which restrict production of anti-tumor factors, such as NO, TNF-α, and IFN-γ, and reprogram macrophages towards the M2 pro-tumor phenotype [13]. Furthermore, tumor cells deteriorate the antigen-presenting function of macrophages [14–16]. As a consequence, macrophages that migrate to the tumor area specifically to destroy tumor cells become tumor “team-mates”. The M2 phenotype of TAM (TAM-M2) suppresses the anti-tumor immunity, promotes growth and vascularization of the tumor, as well as tumor cell invasion and cancer dissemination [1–3]. Indeed, according to clinical and experimental data, the amount of TAM-M2s in the tumor area correlates with a poor prognosis during tumor growth [1,17]. Recently, the concept of a continuum has been proposed [18]. This concept implies that the TAM phenotype varies along a continuous proinflammatory spectrum, in a range from M1 (anti-tumour) to M2 (pro-tumour).
Based on these data, we suggested that M1 macrophages accumulating in the tumor area could limit carcinogenesis and extend the lifespan of the tumor host. The aim of the study was to verify this hypothesis with respect of Ehrlich ascites carcinoma (EAC). The objectives of the study were to evaluate effects of 1) EAC on a macrophage phenotype and NO-producing activity of macrophages in vivo, 2) ascitic fluid from mice with EAC on a macrophage phenotype and NO-producing activity of macrophages in vitro, and 3) macrophages reprogrammed in vitro towards the M1 phenotype and activated with LPS on the lifespan of mice with EAC.
Material and Methods
Experimental animals
Experiments were performed on C57BL/6J mice. Mice were obtained from the vivarium Andreevka (Moscow, Russia) (http://andreevka.msk.ru/index.htm). All experiments were designed and performed in accordance with the WHO guidance for biomedical research in animals (http://www.cioms.ch/publications/guidelines/1985_texts_of_guidelines.htm). The protocol of experiments was approved by the University Ethics Committee. All mice in experimental groups died because of tumor progression.
EAC
Antitumor properties of reprogrammed macrophages were evaluated on an experimental model of Ehrlich ascites carcinoma (EAC). The EAC model was developed in 1905 by transplantation of spontaneous mouse mammary carcinoma [19]. The ascites variant of this strain was obtained in 1933 by transplantation of the subcutaneous strain into the abdominal cavity. Tumor growth was initiated by an intraperitoneal injection of EAC cells. EAC cells were obtained from the N.N. Blokhin Russian Cancer Research Center. Mice were injected intraperitoneally with 250,000 EAC cells diluted in 0.2 ml of saline. The EAC growth curve consisted of 3 periods, including a lag phase (1–5 days after EAC cells injection), log phase (6–10 days), and terminal period (11–15 days) followed by death of the tumor host [19]. All mice were weighed daily until their death. Resistance of mice to EAC was assessed by survival time after the EAC cell injection and changes in the animal weight, which reflected ascitic fluid accumulation in the peritoneal cavity, cachexia and muscle wasting.
Determination of the ascitic fluid EAC cells to macrophages ratio (EAC cells/macrophages ratio) in peritoneal cavity
Ascitic fluid was withdrawn from mice with a syringe on the day of EAC cell injection and on the 11th day after the injection. Cells isolated from the ascitic fluid were counted using a hemocytometer. The cells were placed in a 24-well plate at 1000 cells/ml/well and incubated for one hour at 37.5°C and 5% CO2. Over this period, all macrophages, as distinct from tumor cells, became firmly fixed to the bottom of the wells. The plate was then vortexed, the content of the wells was pipetted and all supernatant was collected. Cells were counted in the supernatant. Cells of the supernatant were represented mainly by tumor cells which did not adhere to the plastic. The EAC cells/macrophages ratio was calculated by formula: (quantity of EAC cells)/(1000-quantity of EAC cells)”.
Assessment of changes in macrophage phenotype and secretory activity
Peritoneal macrophages were isolated from mice [20] on the 11th day after the EAC cells injection to evaluate the effect of EAC on the macrophage phenotype and secretory activity. After isolation, macrophages were placed into wells of flat-bottomed 48-well culture plates containing RPMI-1640 medium with 10% serum (FBS) with 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C and 5% CO2. Macrophages were dispensed into wells containing 0.5 ml of culture medium each at 0.5×106 cells per well. Secretory activity of macrophages was assessed by NO production. For evaluation of basal NO production, macrophages were cultured for 24 hours; for evaluation of stimulated NO production, lipopolysaccharide (LPS) 500 ng/ml was added for 24 hours. Concentration of nitrite, a stable NO metabolite and an index of NO production, was measured in macrophage culture medium [21]. Nitrite was measured spectrophotometrically using the Griess reaction [22].
Shifts of macrophage phenotype were assessed by changes in NO production as well as by amounts of CD80, a cell-surface marker of M1 phenotype, and CD206, a marker of M2 phenotype [23]. The CD markers were measured by flow cytometry using monoclonal antibodies to CD80 (Beckman Coulter, cat # 12-0801-82, PE) and CD206 (Beckman Coulter, cat # FAB2535P, PE) [24] according to the manufacturer’s instructions. The amount of macrophages with M1 and M2 CD markers was expressed in per cent of total macrophage number. Increased levels of NO and M1 phenotype CD markers indicated formation of the M1 phenotype, whereas decreases in these indices indicated formation of the M2 phenotype [23].
Evaluating the effect of ascitic fluid on secretory activity of cultured macrophages
Ascitic fluid was collected into a syringe on 11th day after the EAC cells injection. Concentration of nitrite in ascitic fluid was measured spectrophotometrically using the Griess reaction [22]. The macrophage culture medium was then replaced with the ascitic fluid, and basal and stimulated macrophage NO production was assessed.
Evaluating the effect of EAC cells on secretory activity of cultured macrophages
EAC cells were added to the culture medium at 500×103. Basal and stimulated macrophage NO production was evaluated as described above. Since in the culture medium and the medium after culturing tumor cells (without macrophages), the nitrite concentration was below the quantification limit (less than 2–3 μmol/l), it is reasonable to consider the changes in co-culture nitrite reflecting changes in macrophage NO production.
Methods of in vitro macrophage reprogramming towards M1 phenotype and activating with LPS
Macrophages were reprogrammed towards the M1 phenotype using two reprogramming factors, 0% FBS [25] and 20 ng/ml IFN-γ [22]. Macrophages were isolated from intact mice and divided into four groups.
Macrophages cultured for 36 hours under the standard conditions with 10% FBS (non-reprogrammed and non-stimulated macrophages).
Macrophages cultured for 36 hours without FBS, with IFN-γ 20 ng/ml (reprogrammed in vitro towards M1 phenotype and non-stimulated macrophages).
Macrophages cultured for 12 hours under the standard conditions with 10% FBS and then stimulated with LPS (500 ng/ml) for 24 hours (non-reprogrammed and stimulated with LPS macrophages).
Macrophages incubated for 12 hours without FBS, with IFN-γ 20 ng/ml and then stimulated with LPS (500 ng/ml) for 24 hours (reprogrammed in vitro towards M1 phenotype and stimulated with LPS macrophages).
To confirm the effectiveness of macrophage reprogramming towards the M1 phenotype, we measured NO production, pro- and anti-inflammatory cytokines, and CD-markers of the phenotype. Concentrations of the M1 proinflammatory cytokines, IL-2, IL-6, IFNγ, TNF-α, and the M2 anti-inflammatory cytokines, IL-5 and IL-10 [19,26,27], were determined by flow cytometry (Beckman Coulter FC500, USA) using a specific multiplex kit (BMS810FF, BenderMedSystems, USA) according to the manufacturer’s instructions.
Methods of removing macrophages from the plates and injecting into the peritoneal cavity of mice
Macrophages from Groups 1, 2, 3, and 4 were removed from the bottom of culture wells using trypsin [28] and injected intraperitoneally to intact and EAC mice. The culture medium was aspirated from the wells and then 1 ml of 0.25% trypsin solution with 0.03% EDTA was added to the wells. Plates were incubated at 37°C for 3 min. Then plates were shaken up. One ml of culture medium was then added to each well and the plate was shaken up. The liquid from the wells was decanted into tubes, and 1 ml of culture medium was added to each well again. The plate was shaken up once more and the liquid was placed into the same tube. The tubes were centrifuged for 5 min at 1000 rpm. Then the supernatant was removed from the tube. Cell pellet was resuspended with 1 ml of culture medium and pipetted to obtain a suspension of macrophages. The macrophage concentration was adjusted to 1×106 cells per 1 ml of culture medium with the RPMI-1640 medium (solution A).
Solution A was used to prepare a macrophage suspension at a concentration of 4×106 cells in 0.5 ml PBS (solution B). Each mouse was injected with solution B intraperitoneally on days 3, 7 and 11 after the EAC cell injection. We formed 5 groups each containing 16 mice:
”Tumor” group, mice injected with EAC cells;
“Tumor + PBS” group, mice injected with EAC cells with subsequent infusion of PBS 0.5 ml at 3, 7 and 11 days;
“Tumor + M0-Mac” group, mice injected with EAC cells with subsequent injections of a suspension of 4×106 non-reprogrammed and stimulated with LPS macrophages in 0.5 ml of PBS on days 3, 7 and 11 of the experiment;
“Tumor + M1-Mac” group, mice injected with EAC cells with subsequent injections of a suspension of 4×106 reprogrammed in vitro towards M1 phenotype and stimulated with LPS macrophages in 0.5 ml of PBS on days 3, 7 and 11 of the experiment;
“Tumor + cisplatin” group, mice injected with EAC cells with subsequent infusion of 0.05 ml of cisplatin (0.5 mg/ml), an antitumor drug [29,30], on days 3, 7 and 11 experimental days. Cisplatin has been often used as a reference drug [31].
The effect of the injected macrophages and cisplatin was evaluated based on changes in body weight and lifespan of mice with EAC.
Statistical analyses were performed using analysis of variance followed by Student-Newman-Keuls test. Data are presented as mean (M) with standard errors of the mean (±SEM). Differences were considered statistically significant at p<0.05.
Results
Changes in the phenotype and secretory activity of macrophages in mice with EAC
After tumor cells emerge in the body, two processes are initiated at the same time, tumor cells division and migration of macrophages to the tumor growth area. These processes result in increased amount of tumor cells and macrophages. We found that after the tumor cell injection into mice, the division rate of tumor cells exceeded the rate of macrophage migration to the tumor area. In result, on the day of EAC cell injection, the ratio of EAC cells to macrophage number in the peritoneal cavity was 0.5, and on day 11 it was 1.5 (Table 1). Table 1 shows that the increases in the tumor cells/macrophages ratio in EAC are associated with a 50% decrease in nitrite concentration in the peritoneal cavity and inhibition of the macrophage capability for synthesizing NO both in the basal conditions and under the LPS stimulation. In the terminal phase (day 11) of EAC, the basal NO macrophage production (estimated by nitrite concentration) was decreased 3-fold whereas the stimulated production was decreased 4-fold as compared with the day of EAC cell injection (day 0).
Table 1.
Changes in peritoneal/ascitic fluid nitrite concentration, NO-producing activity of macrophages, and surface CD markers of macrophage phenotype in mice with EAC.
| Days after tumor cell injection | ||
|---|---|---|
| 0 | 11 | |
| Nitrite concentration in mouse peritoneal/ascitic fluid, μmol/l | 65.8±5.7 | 28.7±0.7 |
| Basal NO production (nitrite, μmol/l) | 17±1.9 | 5±0.3** |
| Stimulated NO production (nitrite, μmol/l) | 25±2.1 | 6±0.4** |
| CD marker of M1 phenotype, CD80, % | 80.6±5.6 | 67.7±4.3* |
| CD marker of M2 phenotype, CD206, % | 52.7±4.9 | 89.4±7.9* |
| Tumor cells/macrophages ratio in peritoneal cavity | 0.5 | 1.5 |
Statistical significance vs. control (day 0):
p<0.01;
p<0.05.
Low NO production is a marker of pro-tumor, M2 phenotype [23]. Therefore, we can conclude that EAC shifts the murine macrophage phenotype towards the M2, pro-tumor phenotype. The pro-tumor shift of phenotype is confirmed by CD markers of the macrophage phenotype. At the end stage of EAC, macrophages expressed more CD markers of the M2 phenotype, CD206, and less markers of the M1 phenotype, CD80, as compared with macrophages from mice without EAC (Table 1). The decline of ascitic nitrite concentration during the EAC growth might also reflect reduced NO-producing activity of the M2 phenotype.
Effects of ascitic fluid and EAC cells on secretory activity of cultured macrophages
Reprogramming of EAC mice peritoneal macrophages towards the M2 phenotype may occur under the influence of various ascitic factors and EAC cells. We separately evaluated the effect of ascitic fluid and EAC cells on secretory activity of cultured macrophages isolated from mice without EAC. The results are presented in Table 2. Ascitic fluid and EAC cells inhibited the NO-producing activity of cultured macrophages (estimated by nitrite concentration). Thus, the basal NO production decreased by 50% and the stimulated NO production – by 20% under the action of ascitic fluid. EAC cells decreased the basal NO production by 27% and the stimulated NO production by 29%.
Table 2.
Effect of ascitic fluid isolated from mice with EAC and of EAC cells on NO production in cultured macrophages.
| Nitrite concentration in cell culture (μmol/l) | ||
|---|---|---|
| Basal conditions | Stimulated conditions | |
| Macrophages, without ascitic fluid and EAC cells | 19.1±1.1 | 23.2±0.5 |
| Macrophages, after addition of ascitic fluid | 9.4±0.7** | 18.9±0.9* |
| Macrophages, after addition of EAC cells | 13.9±0.9* | 16.5±1.1** |
Statistical significance vs. control:
p<0.05,
p<0.01.
Thus, factors soluble in ascitic fluid from EAC mice and EAC cells contribute to macrophage reprogramming towards the M2 phenotype and decrease the NO-producing activity of macrophages.
In vitro macrophage reprogramming towards M1 phenotype
Results of this study provided above showed that EAC macrophages reprogrammed towards the M2 phenotype significantly reduced their NO-producing activity in vivo whereas the ascitic fluid and EAC cells exerted the same effect in vitro. These results suggested that M1 macrophages with high NO-producing activity injected into mice with EAC would limit the EAC growth.
Figure 1 shows changes in the nitrite concentration in a culture medium after in vitro reprogramming towards the M1 phenotype. Macrophages cultured in the absence of FBS but in the presence of added INF-γ both increased the basal nitrite concentration (Figure 1, Basal) and significantly increased the nitrite production response to LPS (Figure 1, LPS). These changes reflect formation of the M1 phenotype with typical, high NO-generating activity. Thus, by removing FBS and adding IFN-γ, we have successfully reprogrammed the functional phenotype of murine macrophages towards the M1 phenotype.
Figure 1.
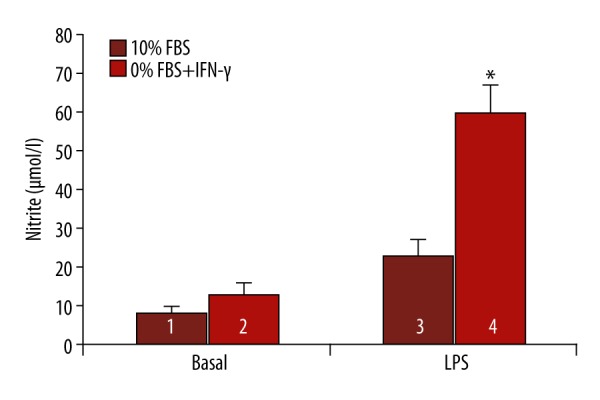
Macrophage reprogramming by 0% FBS and IFN-γ. 1 – non-reprogrammed and non-stimulated macrophages; 2 – reprogrammed in vitro towards M1 phenotype and non-stimulated macrophages; 3 – non-reprogrammed and stimulated with LPS macrophages; 4 – reprogrammed in vitro towards M1 phenotype and stimulated with LPS macrophages. Significant differences between groups 3 and 4: * P<0.01.
To confirm the effectiveness of macrophage reprogramming we also evaluated the secretory activity of LPS-stimulated macrophages by cytokine production. Macrophage reprogramming towards the M1 phenotype resulted in reduced production of the anti-inflammatory cytokines, IL-5 and IL-10, and increased production of the proinflammatory cytokines, IL-2, IL-6, TNF-α and INF-γ, as compared with the cytokine production by non-reprogrammed macrophages (Table 3). All these changes in cytokine levels were statistically significant (p<0.001). The most noticeable effects of reprogramming were an almost 3.5-fold increase in the level of the proinflammatory cytokine, IL-2, and an almost 50% decrease in the level of the anti-inflammatory cytokine, IL-5 (Table 3). Such change in the cytokine production is a feature of macrophage reprogramming towards the M1 phenotype [32]. Secretory activity specifically of LPS-activated macrophages was evaluated for two reasons. First, phenotypic differences are most evident in activated macrophages (e.g., see Figure 1); and second, these were, specifically, LPS-activated, reprogrammed macrophages that were injected into mice with tumor to restrict the tumor growth.
Table 3.
Effect of reprogramming (0%FBS + IFN-γ) on macrophage production of pro- and anti-inflammatory cytokines.
| Cytokines | Non-reprogrammed macrophages + LPS | Reprogrammed macrophages + LPS |
|---|---|---|
| IL-2, M1 | 546.74±67.20 | 1894.72±351.2* |
| IFN-γ, M1 | 8602.36±1045.91 | 14639.49±684.55* |
| TNF-α, M1 | 3548.06±733.62 | 9675.58±647.65* |
| IL-6, M1 | 7645.63±748.86 | 15240.60±872.30* |
| IL-5, M2 | 132.45±41.48 | 67.80±8.45* |
| IL-10, M2 | 468.25±81.85 | 272.21±12.47* |
| CD markers of phenotype (%) | ||
| CD marker of M1 phenotype, CD80 | 6.4±1.4 | 12.1±2.2** |
| CD marker of M2 phenotype, CD206 | 38.0±2.7 | 27.6±2.3** |
Statistical significance vs. non-reprogrammed macrophages + LPS:
p<0.001;
p<0.001.
The phenotype shift towards M1 was confirmed by changes in expression of CD markers. After reprogramming, macrophages expressed more CD80, the M1 phenotype surface marker, and less CD206, the M2 phenotype marker, than macrophages before reprogramming (Table 3).
Thus, the analysis of macrophage phenotype markers, such as NO and cytokine production and expression of surface CD markers, allowed us to consider the suggested model as an appropriate in vitro model of macrophage reprogramming towards the M1 phenotype.
Macrophages reprogrammed in vitro towards the M1 phenotype and activated with LPS extend lifespan of mice with EAC
Figure 2 shows effects of injected macrophages and cisplatin on lifespan of mice with EAC in Kaplan-Meier survival plots. The survival duration of mice injected with EAC cells was 13.6±0.2 days (“Tumor” group) whereas the survival duration of mice injected with macrophages reprogrammed towards the M1 phenotype and stimulated with LPS (“Tumor + M1-Mac” group) was 22.8±0.8 days (p<0.01), i.e., 68% longer than in the “Tumor” group. Survival duration of mice injected with macrophages reprogrammed towards the M1 phenotype and stimulated with LPS was longer than survival duration of mice injected with the antitumor drug cisplatin («Tumor + cisplatin» group).
Figure 2.
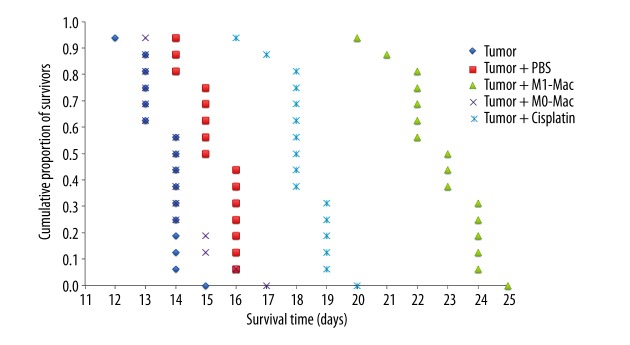
The effect of the injected macrophages and cisplatin on lifespan of mice with EAC in Kaplan-Meier survival plots. “Tumor” group, mice injected with EAC cells; “Tumor + PBS” group, mice injected with EAC cells with subsequent infusion of PBS 0.5 ml at 3, 7 and 11 days; “Tumor + M0-Mac” group, mice injected with EAC cells with subsequent injections of a suspension of 4×106 non-reprogrammed and stimulated with LPS macrophages in 0.5 ml of PBS on days 3, 7 and 11 of the experiment; “Tumor + M1-Mac” group, mice injected with EAC cells with subsequent injections of a suspension of 4×106 reprogrammed in vitro towards M1 phenotype and stimulated with LPS macrophages in 0.5 ml of PBS on days 3, 7 and 11 of the experiment; “Tumor + cisplatin” group, mice injected with EAC cells with subsequent infusion of 0.05 ml of cisplatin (0.5 mg/ml), an antitumor drug, on days 3, 7 and 11 experimental days.
Infusion of PBS (“Tumor + PBS” group) or non-reprogrammed, LPS-stimulated macrophages (“Tumor + M0-Mac” group) did not significantly influence the survival duration in mice with EAC.
The toxic effect of EAC in all groups was evident as progressive ascitic fluid accumulation in peritoneal cavity and, therefore, increasing body weight of mice. However, in mice injected with macrophages reprogrammed towards the M1 phenotype and stimulated with LPS (“Tumor + M1-Mac” group), the body weight increased by 6.5±1.1% at 11 days of EAC development from the 1st day, whereas in mice not injected with macrophages (“Tumor” group), the body weight increased by 12.7±1.8% (p<0.01), i.e., twice as much as in macrophage-treated mice.
Figure 3 shows mice on day 11 after the EAC cell injection. It is obvious that peritoneal accumulation of ascitic fluid was significantly less severe in mice injected with macrophages reprogrammed towards the M1 phenotype and stimulated with LPS than in mice not injected with macrophages.
Figure 3.
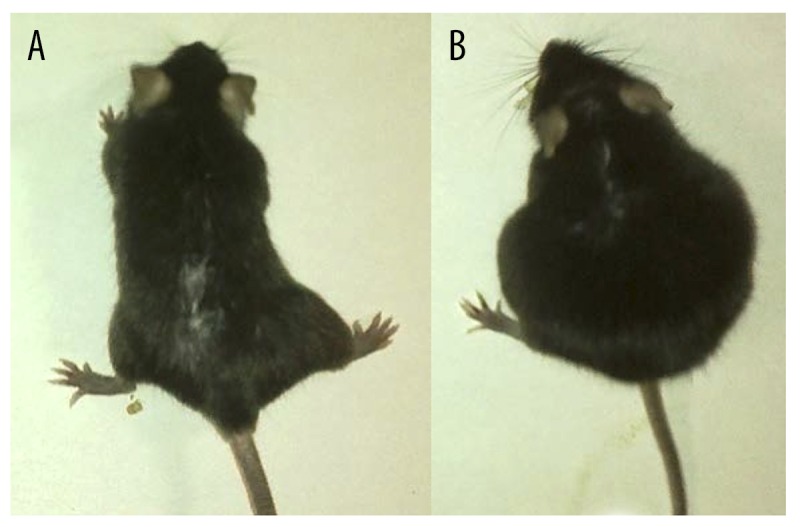
Mice with EAC injected and not injected with reprogrammed in vitro towards M1 phenotype and stimulated with LPS macrophages. (A) Mice injected with reprogrammed in vitro towards M1 phenotype and stimulated with LPS macrophages (“Tumor + M1-Mac” group); (B) Mice not injected with macrophages (“Tumor” group).
Infusion of PBS (“Tumor + PBS” group) or non-reprogrammed, LPS-stimulated macrophages (“Tumor + M0-Mac” group) did not significantly influence the weight gain of mice with EAC.
Therefore, injection of macrophages reprogrammed in vitro towards the M1 phenotype and stimulated with LPS significantly increased the resistance of mice to development of EAC, which is confirmed by differences in survival duration and peritoneal accumulation of ascitic fluid between mice injected and not injected with macrophages.
Discussion
The experimental EAC model reproduces an important aspect of many human malignancies—accumulation of ascitic fluid in the abdominal cavity. Considerable accumulation of ascitic fluid occurs in ovarian, pancreatic, gastric, and colorectal malignancies [33–35]. Development of these tumors is associated with accumulation in ascitic fluid of large amounts of macrophages [36] that have the M2 phenotype [37], especially the M2 macrophages that play an important role in depression of immune response and tumor cell proliferation [37]. For this reason, reprogramming of tumor-associated macrophages from the protumor M2 to the antitumor M1 phenotype is now beginning to be considered a promising approach to restriction of tumor growth [38,39].
This study demonstrated that the microenvironment, which is formed by experimantal EAC around peritoneal macrophages in vivo, and ascitic fluid or EAC cells added to macrophages in vitro provide macrophage reprogramming towards the M2 phenotype. These findings are consistent with reported data on pro-tumoral effects of the M2 macrophage phenotype [40,41]. At least two groups of ascitic factors participate in the macrophage reprogramming towards the M2 phenotype: first, immune factors, such as the anti-inflammatory cytokine, IL-10 secreted by EAC [42], and, second, physical and chemical factors, such as hypoxia [43] and changed pH [44], which always accompany tumor growth [45].
The main result of the study was that the macrophages reprogrammed in vitro towards the M1 phenotype and activated by LPS prolonged the lifespan of mice with EAC. The antitumor effect of these macrophages in EAC is consistent with the view that M1 macrophages possess antitumor properties [24,40,41,46].
Our findings and data in the literature allow us to understand mechanisms that restrict EAC growth and extend the lifetime after the injection of exogenous LPS-activated M1 macrophages into the area of tumor growth. First of all, our results confirm well-known data that M1 macrophages have a great capability for NO production. NO plays a dual role in carcinogenesis. On one hand, NO can induce apoptosis of tumor cells by inhibiting synthesis of anti-apoptotic Bcl-2 and increasing expression of proapoptotic Bax and p53 [4], and, thereby, exerting anti-tumor effects [47,48]. However, on the other hand, NO can hamper development of apoptosis in tumor cells and exert a pro-tumoral effect due to caspase nitrosylation [49,50] and/or activation of HSP70 synthesis [51,52]. Local concentrations of NO and free radicals in the tumor zone determine the interaction between NO and its metabolites and DNA, DNA repair systems, the tumor suppressor, p53, and other activators and inhibitors of tumor cell apoptosis. These interactions, in turn, will determine pro- or antitumor effects of NO [53]. In our study, the injection of LPS-activated M1 macrophages with high NO-producing activity extended the survival time of mice with EAC. Therefore, in this case, we observed an anti-tumor effect of NO.
Activated M1 macrophages can limit tumor cell growth, not only through NO production, but also through other mechanisms, such as increased production of pro-inflammatory cytokines [7,48]; free radicals [47]; activation of natural killer cells, which can effectively destroy tumor cells [54,55]; and presentation of tumor antigens and formation of Th1 and cytotoxic lymphocytes [56,57], which infiltrate the tumor and kill tumor cells [9]. Indeed, in our experiments, reprogrammed M1 macrophages produced more proinflammatory cytokines, including IL-2, IFN-γ, and TNF-α, which possess well-described antitumor properties, and significantly less L-5 and IL-10, which possess protumor properties, compared to non-reprogrammed macrophages [58].
At present, we cannot determine which of the above-mentioned mechanisms mediates the effect of exogenous LPS-activated M1 macrophages on the EAC growth limitation. However, the substantial prolongation of mouse lifetime after an injection of these macrophages suggests that promising biotechnologies for restriction of tumor growth could be developed based on the in vitro macrophage reprogramming. The promising nature of this direction was demonstrated for the first time by Andreesen et al. [59] and Allavena et al. [60], and then confirmed in studies of Hagemann et al. [61] and Coscia et al. [62].
Conclusions
EAC reprograms macrophages towards the pro-tumor M2 phenotype and significantly decreases their NO-producing activity. Culturing without FBS but with added INF-γ reprograms macrophages towards the M1 phenotype and significantly increases the basal and stimulated macrophage NO production. Macrophages reprogrammed in vitro towards the M1 phenotype and activated by LPS extend the survival duration of mice with EAC. This effect was superior to that of the known antitumor drug, cisplatin.
Footnotes
Source of support: This study was conducted with governmental financial support represented by the Ministry of Education and Science of the Russian Federation (Agreement dated June 17, 2014, No. 14.604.21.0020. The unique identifier of applied research: RFMEFI60414X0020)
References
- 1.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–66. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–12. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 3.Sica A, Schioppa T, Mantovani A, Allavena P. Tumor-associated macrophages are a distinct M2 polarized population promoting tumor progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–27. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Zeini M, Través PG, López-Fontal R, et al. Specific contribution of p19(ARF) to nitric oxide-dependent apoptosis. J Immunol. 2006;177(5):3327–36. doi: 10.4049/jimmunol.177.5.3327. [DOI] [PubMed] [Google Scholar]
- 5.Harris DP, Haynes L, Sayles PC, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol. 2000;1:475–82. doi: 10.1038/82717. [DOI] [PubMed] [Google Scholar]
- 6.Ibe S, Qin Z, Schuler T, et al. Tumor rejection by disturbing tumor stroma cell interactions. J Exp Med. 2001;194:1549–59. doi: 10.1084/jem.194.11.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsung K, Dolan JP, Tsung YL, Norton JA. Macrophages as effector cells in interleukin 12-induced T cell-dependent tumor rejection. Cancer Res. 2002;62:5069–75. [PubMed] [Google Scholar]
- 8.Sharma M. Chemokines and their receptors: orchestrating a fine balance between health and disease. Crit Rev Biotechnol. 2010;30(1):1–22. doi: 10.1080/07388550903187418. [DOI] [PubMed] [Google Scholar]
- 9.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 10.Elgert KD, Alleva DG, Mullins DW. Tumor-induced immune dysfunction: the macrophage connection. J Leukoc Biol. 1998;64:275–90. doi: 10.1002/jlb.64.3.275. [DOI] [PubMed] [Google Scholar]
- 11.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou W. Regulatory T cells, tumor immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 13.Stout RD, Watkins SK, Suttles J. Functional plasticity of macrophages: in situ reprogramming of tumor-associated macrophages. J Leukoc Biol. 2009;86(5):1105–9. doi: 10.1189/jlb.0209073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabrilovich D. Mechanisms and functional significance of tumor-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–52. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 15.Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000;74:181–273. doi: 10.1016/s0065-2776(08)60911-6. [DOI] [PubMed] [Google Scholar]
- 16.Seliger B, Maeurer MJ, Ferrone S. Antigen-processing machinery breakdown and tumor growth. Immunol Today. 2000;21:455–64. doi: 10.1016/s0167-5699(00)01692-3. [DOI] [PubMed] [Google Scholar]
- 17.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–40. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ostrovskaia LA, Skibida IP, Krugliak SA, Emanuél’ NM. Kinetic peculiarities of the development of Ehrlich ascites tumor in linear and non-linear mice. Izv Akad Nauk SSSR Biol. 1966;5:734–38. [PubMed] [Google Scholar]
- 20.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14(Unit 14):1. doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lancaster JR. Simulation of the diffusion and reaction of endogenously produced nitric oxide. Jr Proc Natl Acad Sci USA. 1994;91(17):8137–41. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Redente EF, Dwyer-Nield LD, Barrett BS, et al. Lung tumor growth is stimulated in IFN-gamma−/− mice and inhibited in IL-4Ralpha−/− mice. Anticancer Res. 2009;29(12):5095–101. [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;1(13):453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 24.Hao NB, Lü MH, Fan YH, et al. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–95. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mills CD, Kincaid K, Alt JM, et al. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–73. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 26.Diehl S, Rincón M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol. 2002;39(9):531–36. doi: 10.1016/s0161-5890(02)00210-9. [DOI] [PubMed] [Google Scholar]
- 27.Liao W, Lin JX, Leonard WJ. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol. 2011;23(5):598–604. doi: 10.1016/j.coi.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rey-Giraud F, Hafner M, Ries CH. In vitro generation of monocyte-derived macrophages under serum-free conditions improves their tumor promoting functions. PLOS One. 2012;7(8):e42656. doi: 10.1371/journal.pone.0042656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nan Zhao, Li YH, Wu XK, et al. Effect of Brucea javanica fruit oil emulsion combined cisplatin on the growth inhibition of transplanted tumor in human ovarian cancer SKOV3 nude mice: an experimental study. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2015;35(1):57–62. [PubMed] [Google Scholar]
- 30.Roco A, Cayún J, Contreras S, et al. Can pharmacogenetics explain efficacy and safety of cisplatin pharmacotherapy? Front Genet. 2014;5:391. doi: 10.3389/fgene.2014.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen TC, Cho HY, Wang W, et al. Chemotherapeutic effect of a novel temozolomide analog on nasopharyngeal carcinoma in vitro and in vivo. J Biomed Sci. 2015;22(1):71. doi: 10.1186/s12929-015-0175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–95. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cavazzoni E, Bugiantella W, Graziosi L, et al. Malignant ascites: pathophysiology and treatment. Int J Clin Oncol. 2013;18:1–9. doi: 10.1007/s10147-012-0396-6. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed N, Stenvers KL. Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Front Oncol. 2013;3:256. doi: 10.3389/fonc.2013.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saif MW, Siddiqui IA, Sohail MA. Management of ascites due to gastrointestinal malignancy. Ann Saudi Med. 2009;29:369–77. doi: 10.4103/0256-4947.55167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Deavers M, Patenia R, et al. Monocyte macrophage and T-cell infiltrates in peritoneum of patients with ovarian cancer or benign pelvic disease. J Transl Med. 2006;4:30. doi: 10.1186/1479-5876-4-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi K, Komohara Y, Tashiro H, et al. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Sci. 2010;101:2128–36. doi: 10.1111/j.1349-7006.2010.01652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Na YR, Yoon YN, Son DI, Seok SH. Cyclooxygenase-2 inhibition blocks M2 macrophage differentiation and suppresses metastasis in murine breast cancer model. PLoS One. 2013;8:e63451. doi: 10.1371/journal.pone.0063451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kono Y, Kawakami S, Higuchi Y, et al. Antitumor effect of nuclear factor-κB decoy transfer by mannose-modified bubble lipoplex into macrophages in mouse malignant ascites. Cancer Sci. 2014;105(8):1049–55. doi: 10.1111/cas.12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 2014;6(3):1670–90. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galdiero MR, Bonavita E, Barajon I, et al. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013;218(11):1402–10. doi: 10.1016/j.imbio.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 42.da Silva RJ, da Silva MG, Vilela LC, Fecchio D. Cytokine profile of Ehrlich ascites tumor treated with Bothrops jararaca venom. Mediators Inflamm. 2002;11(4):197–201. doi: 10.1080/0962935029000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hao NB, Lü MH, Fan YH, et al. Macrophages in tumor microenvironments and the progression of tumors. Dev Immunol. 2012;2012:948098. doi: 10.1155/2012/948098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lardner A. The effects of extracellular pH on immune function. J Leukoc Biol. 2001;69(4):522–30. [PubMed] [Google Scholar]
- 45.Kumar V, Gabrilovich DI. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunol. 2014;143(4):512–19. doi: 10.1111/imm.12380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mills CD, Thomas AC, Lenz LL, Munder M. Macrophage: SHIP of Immunity. Front Immunol. 2014;5:620. doi: 10.3389/fimmu.2014.00620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lum HD, Buhtoiarov IN, Schmidt BE, et al. Tumoristatic effects of anti-CD40 mAb-activated macrophages involve nitric oxide and tumour necrosis factor-α. Immunol. 2006;118(2):261–70. doi: 10.1111/j.1365-2567.2006.02366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maejima Y, Adachi S, Morikawa K, et al. Nitric oxide inhibits myocardial apoptosis by preventing caspase-3 activity via S-nitrosylation. J Mol Cell Cardiol. 2005;38(1):163–74. doi: 10.1016/j.yjmcc.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 50.Török NJ, Higuchi H, Bronk S, Gores GJ. Nitric oxide inhibits apoptosis downstream of cytochrome C release by nitrosylating caspase 9. Cancer Res. 2002;62(6):1648–53. [PubMed] [Google Scholar]
- 51.Malyshev IY, Malugin AV, Golubeva LY, et al. Nitric oxide donor induces HSP70 accumulation in the heart and in cultured cells. FEBS Lett. 1996;391(1–2):21–23. doi: 10.1016/0014-5793(96)00691-6. [DOI] [PubMed] [Google Scholar]
- 52.Murphy ME. The HSP70 family and cancer. Carcinogenesis. 2013;34(6):1181–88. doi: 10.1093/carcin/bgt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Engin AB. Dual function of nitric oxide in carcinogenesis, reappraisal. Curr Drug Metab. 2011;12(9):891–99. doi: 10.2174/138920011797470092. [DOI] [PubMed] [Google Scholar]
- 54.Smyth MJ, Thia KY, Street SE, et al. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000;192:755–60. doi: 10.1084/jem.192.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Street SE, Cretney E, Smyth MJ. Perforin and interferon-g activities independently control tumor initiation, growth, and metastasis. Blood. 2001;97:192–97. doi: 10.1182/blood.v97.1.192. [DOI] [PubMed] [Google Scholar]
- 56.Backer R, Schwandt T, Greuter M, et al. Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc Natl Acad Sci USA. 2010;107(1):216–21. doi: 10.1073/pnas.0909541107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barrio MM, Abes R, Colombo M, et al. Human macrophages and dendritic cells can equally present MART-1 antigen to CD8(+) T cells after phagocytosis of gamma-irradiated melanoma cells. PLoS One. 2012;7(7):e40311. doi: 10.1371/journal.pone.0040311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang T, Zhou C. The past, present and future of immunotherapy against tumor. Transl Lung Cancer Res. 2015;4(3):253–64. doi: 10.3978/j.issn.2218-6751.2015.01.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andreesen R, Scheibenbogen C, Brugger W, et al. Adoptive transfer of tumor cytotoxic macrophages generated in vitro from circulating blood monocytes: a new approach to cancer immunotherapy. Cancer Res. 1990;50:7450–56. [PubMed] [Google Scholar]
- 60.Allavena P, Peccatori F, Maggioni D, et al. Intraperitoneal recombinant gamma-interferon in patients with recurrent ascitic ovarian carcinoma: modulation of cytotoxicity and cytokine production in tumor-associated effectors and of major histocompatibility antigen expression on tumor cells. Cancer Res. 1990;50:7318–23. [PubMed] [Google Scholar]
- 61.Hagemann T, Lawrence T, McNeish I, et al. “Re-educating” tumor-associated macrophages by targeting NF-κB”. J Exp Med. 2008;205:1261–68. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coscia M, Quaglino E, Iezzi M, et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J Cell Mol Med. 2010;14:2803–15. doi: 10.1111/j.1582-4934.2009.00926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]