

Summary
Purpose
Triciribine phosphate is a potent, small-molecule inhibitor of activation of all three isoforms of AKT in vitro. AKT is an intracellular protein that, when activated, leads to cellular division; it is dysregulated in a large number of malignancies, and constitutively activating AKT mutations are present in a minority of cancers.
Patients and methods
In this phase I study triciribine phosphate monohydrate (TCN-PM) was administered to subjects whose tumors displayed evidence of increased AKT phosphorylation (p-AKT) as measured by immunohistochemical analysis (IHC). TCN-PM was administered over 30 min on days 1, 8 and 15 of a 28-day cycle. Tumor biopsy specimens, collected before treatment and on day +15, were assessed for p-AKT by IHC and western blot analyses.
Results
Nineteen subjects were enrolled; 13 received at least one cycle of therapy, and a total of 34 complete cycles were delivered. One subject was treated at the 45 mg/m2 dose before the study was closed due to its primary objective having been met. No dose-limiting toxic effects were observed. Modest decreases in tumor p-AKT following therapy with TCN-PM were observed at the 35 mg/m2 and 45 mg/m2 dose levels, although definitive conclusions were limited by the small sample size.
Conclusions
These preliminary data suggest that treatment with TCN-PM inhibits tumor p-AKT at doses that were tolerable. Although single agent activity was not observed in this enriched population, further combination studies of TCN-PM with other signal transduction pathway inhibitors in solid tumors is warranted.
Keywords: AKT, Triciribine, Phosphorylation, Phase I
Introduction
AKT (also known as protein kinase B) is a serine/threonine protein kinase and is the human homolog of the viral oncogene v-akt [1]. Three human isoforms exist: AKT1/PKBα, AKT2/PKBβ and AKT3/PKBγ. Stimulation of cells with growth or survival factors results in recruitment of a lipid kinase phosphoinositide-3-OH-kinase (PI3K) to cell surface receptors. Activated PI3K phosphorylates phosphoinositol-4, 5-biphosphate (PIP2) to phosphoinositol-3,4,5-biphosphate (PIP3), which recruits AKT to the plasma membrane, where it can be activated by phosphorylation on a threonine at position 308 and a serine at position 473 (AKT1), Thr308 and Ser474 (AKT2), and Thr308 and Ser472 (AKT3) [2]. The phosphatase and tensin homolog (PTEN) dephosphorylates PIP3 to PIP2 and prevents activation of AKT.
Many human cancers contain hyperactivated AKT, including cancers of the breast, ovary and pancreas [2–4]. This is due to genomic amplification or overexpression of AKT itself, as well as to genetic alterations upstream of AKT, including overexpression of receptor tyrosine kinases or their ligands and mutation or deletion of PTEN [2]. The involvement of AKT in oncogenesis was demonstrated preclinically by showing that ectopic expression of AKT induces malignant transformation and promotes cell survival and that disruption of AKT pathways inhibits cell growth and induces apoptosis [4, 5]. Tumor AKT over-expression and loss of PTEN (all of which result in hyperactivation of AKT) are associated with an adverse prognosis, resistance to chemotherapy, and shortened survival time in cancer patients. Because AKT plays an important part in cell cycle regulation and is frequently dysregulated in human cancers, it is an attractive target for anticancer therapy [6].
Triciribine phosphate (TCN-P, NSC-280594) is a nucleotide derivative first synthesized by Schram and Townsend in 1971 [7]; the chemical structure is depicted in Fig. 1. TCN-P has been tested previously in phase I and II clinical trials, where objective radiographic responses were observed, albeit infrequently (Table 1) [8–13]. Administered as a 120-hour (5-day) infusion every 42 days, the recommended phase II dose was 20 mg/m2/day [8]; pharmacokinetic studies were not performed. The investigators noted cumulative toxicity in the 30 mg/m2/day cohort; the major toxic effects associated with the drug were myelosuppression, elevated pancreatic enzyme levels, hyperglycemia and hepatotoxicity. The agent exhibited activity in a subject with papillary thyroid cancer, and mixed radiographic responses were observed in subjects with soft tissue sarcoma, colorectal cancer or tonsillar carcinoma. When a weekly intravenous regimen was used (days 1, 8, 15 and 22 every 42 days), the highest tolerated dose was 48 mg/m2 [10]. Prolonged retention of the drug was noted, particularly in the liver; drug was detectable in a necropsy specimen from a patient who died 61 days following the last day of treatment. Furthermore, plasma drug levels were unpredictable in nine patients, most likely because of enterohepatic circulation.
Fig. 1.
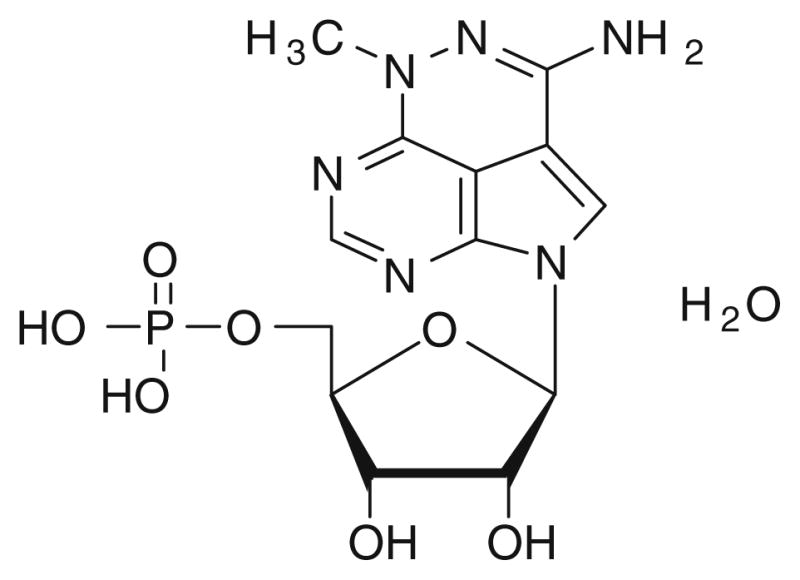
The chemical structure of triciribine phosphate (TCN-P)
Table 1.
Prior Phase I and II studies of TCN-P
| Phase I | ||||
|---|---|---|---|---|
| Author | Dose | MTD | Toxicity | Efficacy |
| Feun [8] | 5-day infusion every 42 d | 20 mg/m2 | Hypertriglyceridemia Hyperamylasemia |
Partial response-1 (thyroid) Stable—1 |
| Mittleman [9] | Intravenous every 21 days | Not reported Treated up to 350 mg/m2 |
Liver dysfunction Hypertriglyceridemai Hypophosphatemia Hypocalcemia |
0/26 |
| Schilcher [10] | Intravenous days 1,8,15, 22 every 42 days | 48 mg/m2 | Thrombocytopenia Anemia Hepatic dysfunction |
0/24 |
| Phase II | ||||
| Tumor type | Dose | Schedule | CR/PR | Toxicity |
| Cervix [11] | 35 mg/m2/d | 5 day infusion q 42 days | 2/21 | Hypocalcemia |
| Breast [12] | 20–40 mg/m2/d | 5 day infusion q 42 days | 0/24 | Asthenia Hypertriglyceridemia |
| Colon [13] | 35 mg/m2 iv | Q 21 days | 1/31 | Hypocalcemia |
MTD maximun tolerated dose, CR complete reponse, PR partial response
The 5-day continuous infusion regimen was studied in a phase II trial of 24 patients with advanced cervical squamous cell carcinoma, at a starting dose of 35 mg/m2/day [11]. Two objective responses were seen, one a complete response lasting greater than 19 months, and another a partial response lasting greater than 5 months. Only one subject developed a grade 4 toxic effect. A phase II study of TCN-P at doses of 20–40 mg/m2 administered as a 24-hour infusion daily for 5 days every 6 weeks yielded no objective responses in 14 subjects with metastatic breast cancer; severe fatigue and hypertriglyceridemia were noted at the 35 mg/m2 and 40 mg/m2 dose levels [12]. The collective conclusion of these referenced studies was that, because of the significant toxicity observed, combined with its lack of significant radiographic activity, further investigation of TCN-P was not warranted.
Small-molecule inhibitors of AKT were identified by evaluating a chemical library of 1,992 compounds from the National Cancer Institute Diversity Set for agents capable of inhibiting growth of AKT2-transformed but not parental NIH/3T3 cells [14]. Of the 32 compounds that selectively inhibited growth of AKT2-transformed cells, the most potent was triciribine. Once inside cells triciribine is converted to TCN-P by adenosine kinase [15]. Triciribine significantly inhibited AKT phosphorylation at both Thr309 and Ser474, which is required for full activation of AKT, and triciribine suppressed epidermal growth factor-induced kinase activity and phosphorylation of all three isoforms of AKT. The kinase activity of recombinant, constitutively active AKT2 (Myr-AKT2) was not inhibited by TCN in an in vitro kinase reaction, suggesting that triciribine does not directly inhibit Akt. Studies have shown that triciribine can selectively inhibit the Akt pathway in vitro; triciribine inhibited growth of cells that overexpress (or have hyperactivated) AKT compared to human cancer cell lines that do not.
Given preclinical data demonstrating that triciribine is a potent intracellular inhibitor of all isoforms of AKT in vivo, we undertook an open-label, phase I dose-escalation study of TCN-P monohydrate (TCN-PM) monotherapy, with pharmacokinetic and pharmacodynamic correlates, restricted to subjects whose tumors had evidence of activated (hyperphosphorylated) AKT. The predetermined objective of the study was to determine whether a reduction in the levels of tumor p-AKT following treatment with TCN-PM could be measured; secondary objectives were to evaluate the safety and pharmacokinetics of a weekly dosing schedule, and to observe any efficacy in patients with solid tumors selected for activated AKT.
Patients and methods
Study design
An open-label phase I study design was used, involving subjects with advanced malignancies refractory to standard therapies, or for which no proven effective therapy existed. A dose-escalation scheme was extrapolated from the previously determined maximum tolerated dose (MTD) of 48 mg/m2 administered on a slightly different weekly schedule (i.e., days 1, 8, 15, and 22, on a 42-day cycle) [10]. Subjects gave written and verbal consent before study entry. The study followed the ethical principles of Good Clinical Practice in accordance with the Declaration of Helsinki. The study was approved by the H. Lee Moffitt Cancer Center Scientific Review Committee and by the University of South Florida Institutional Review Board.
Patient selection
Standard phase I eligibility criteria applied to this study. Eligibility for this study required prior enrollment on a separate tissue study allowing pathological analysis of archival tissue to determine a subject’s tumor p-AKT levels by immunohistochemical analysis (IHC); subjects must have had evidence of tumor AKT hyperphosphorylation to be eligible to participate in this study. All subjects were treated at the H. Lee Moffitt Cancer Center and Research Institute Clinical Research Unit.
Immediately prior to beginning study treatment (days −7 to −1), each patient underwent a tumor biopsies; three to six tru-cut biopsy specimens (either 18 or 20 French gauge) were obtained with image guidance. Only patients with biopsy-confirmed evidence of p-AKT in the tumor (≥ grade 2+ by IHC or a positive results on western blot analysis prior to treatment with study drug were enrolled); absence of p-AKT on tumor sampling by either technique led to study withdrawal.
Drug handling and dispensing
TCN-PM was supplied by Vioquest Pharmaceuticals as lyophilized powder in 50-mg vials. Prior to use, the drug was reconstituted with 2.5 mL of sterile water for a final concentration of 20 mg/mL/vial with a pH of 6.0 to 7.5. The study drug was then diluted in 500 mL isotonic saline solution and infused intravenously over 1 h by means of a regulated infusion pump in the Clinical Research Unit. The research subject’s heart rate and blood pressure were evaluated every 15 min during infusion of the drug. Prophylactic premedication was not required, but because of the agent’s known gastrointestinal toxicity, prophylactic antiemetics were administered at the treating investigator’s discretion. Leukocyte growth factors were not permitted on this study; erythrocyte growth factors were allowed.
Treatment and dose escalation
The starting dose was 15 mg/m2, and was derived by extrapolating from data from prior clinical studies utilizing a slightly different weekly regimen (in that previous study triciribine was administered weekly on days 1, 8, 15, and 22 every 42 days with an MTD of 48 mg/m2); subsequent dose levels were 25 mg/m2, 35 mg/m2 and 45 mg/m2. The weekly schedule was preferred because of the drug’s long terminal half-life (89.2 h) [15]. Intra-patient dose escalation was not allowed. Three subjects were treated at each dose level; provided no dose-limiting toxic effect (DLT) was observed in the first cycle of treatment, the subsequent three subjects were treated at the next higher dose level. Those subjects were not treated at the next higher dose level until all subjects had completed their first cycle of therapy at the lower dose level. Toxic effects were graded by using the Common Terminology Criteria for Toxicity Evaluation version 3.0 (CTCAE v3.0) [16]. A DLT was defined, for the purposes of this study, as any of the following events occurring in the first 4 weeks of study treatment: grade 4 neutropenia (i.e., absolute neutrophil count [ANC] <500 cells/mm3 for 5 or more consecutive days) or febrile neutropenia (i.e., fever >38°C with an ANC <500 cells/mm3 requiring hospitalization); grade 4 thrombocytopenia or bleeding episode requiring platelet transfusion; grade 3 nausea and/or emesis despite the use of maximal medical intervention; grade 2 or greater cardiovascular toxic effect; any grade 3 or greater nonhematologic toxic effect; or delayed recovery (2 weeks or more) after scheduled re-treatment from a delayed toxic effect related to treatment with TCN-PM.
Treatment assessment
Before initiation of therapy, each patient underwent a complete medical history, physical examination, and laboratory evaluation, including complete blood count, electrolyte panel, chemistry group, and urinalysis. Tumor assessment, including serum markers and radiologic assessment of all known measurable sites of disease, was performed prior to initiation of therapy and every 4 weeks. For the first cycle of therapy, an electrocardiogram was performed within 1 h before and again within 1 h after each infusion of TCN-PM. If the corrected QT interval (QTc) lengthened by 60 ms or reached 500 ms, therapy was withheld.
Sample collection and drug levels
Plasma samples (5 mL) were collected in Beckton-Dickinson Vacutainers (or equivalent), which contain K3EDTA as an anticoagulant. Blood samples were collected at time 0 (just prior to TCN-PM infusion) and at 2 h, 24 h, 48 h, 72 h, 96 h and 168 h following completion of infusion (for time points beyond 24 h the times are approximate, plus or minus 2 h). For some patients, an additional optional pharmacokinetic sample was drawn between 120 h and 144 h. Plasma samples were analyzed for concentrations of TCN and TCN-PM by high-performance liquid chromatography.
Pharmacokinetic analysis
Plasma samples were prepared using precipitation extraction with methanol. Chromatography was carried out with an Agilent (Palo Alto, CA) 1100 HPLC system with ultraviolet detection. Elution of both compounds was achieved with a mobile phase of water and acetonitrile, both containing 0.1% TFA. Separation was carried out on a YMC Hydrosphere (Waters, Milford, MA) C18 column with a gradient profile (up to 60% acetonitrile) and a run time of 35 min. Ultraviolet detection was monitored at a wavelength of 320 nm. The method is linear from 5–500 ng/ml for both TCN-PM and TCN. For TCN-PM, inter- and intra-assay variability was 8% and 14%, respectively and contains a relative mean error of less than 7%. TCN presented an inter- and intra-assay variability of 11% and 6% respectively with the assay having a relative mean error of less than 11%. Raw concentrations from patient samples were extrapolated from linear regression applied to calibration samples. The observed Cmax for each compound was recorded along with plotting the concentrations versus time.
Pharmacodynamic analysis
Tumor specimens were acquired by image-guided biopsy as already described and processed (one core in formalin, the remainder flash frozen at −70°C). For IHC analysis, four 1-μm sections were cut with a microtome (Leica Microsystems, Inc., Bannockburn, IL) and transferred to adhesive-coated slides. The slides were dewaxed by heating at 55°C for 30 min and by three washes, 5 min each, with xylene. Tissues were rehydrated by a series of five one-minute washes in 100%, 95%, and 80% ethanol and distilled water. Antigen was retrieved by heating the samples at 95°C for 30 min with 10 mmol/L sodium citrate (pH 6.0). Endogenous peroxidase activity was blocked with 3% hydrogen peroxidase for 20 min. After exposure to universal blocking serum (DAKO Diagnostic, Mississauga, Ontario, Canada) for 30 min, the samples were incubated with monoclonal mouse antiphospho-AKT antibody (clone 587F11; 1:100 dilution; Cell Signaling Technology, Beverly, MA) at 4°C overnight. This antibody identifies the phosphorylated form of AKT at Ser473. The sections were incubated with biotin-labeled secondary antibody and streptavidin-peroxidase for 30 min each (DAKO Diagnostic). The samples were developed with 3,3′-diaminobenzidine substrate (Vector Laboratories, Burlington, Ontario, Canada) and counterstained with hematoxylin. The slides then were dehydrated following standard procedure, and sealed with cover slips. Negative controls were included by omitting p-AKT antibody during primary antibody incubation. The p-AKT IHC slides were examined in a blinded fashion by an expert pathologist (DC).
Positive p-AKT reactions were scored on a scale of four grades according to the intensity of staining: 0, 1+, 2+ and 3+. The percentages of p-AKT–positive cells also were scored on a four-point scale: 0 (0%), 1+ (1–20%), 2+ (21–60%) and 3+ (more than 60%). In cases with a discrepancy between duplicate cores, the higher score was taken as a final score. The product of the intensity and percentage scores was used as the final staining score. The final staining scores were defined as follows: 0, negative; 1 to 3, weak; 4 to 6, moderate; 7 to 9, strong.
For p-Akt analysis by western blotting, core tumor biopsies were lysed in cold TGH buffer (1% Triton X-100, 10% glycerol, 30 mM HEPES pH 7.5) supplemented with 10 mM NaCl and proteinase and phosphatase inhibitors (10 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitor, 25 μg/ml leupeptin, 6.4 mg/ml P-nitrophenyl phosphate, 2 mM phenylmethylsulfonyl fluoride and 2 mM Na3VO4). The Triton-soluble fractions were collected by centrifugation (16,000×g, 20 min) at 4°C. Protein amount was determined using the Bio-Rad Protein Assay reagent, and equal amounts (50 μg) of total protein were loaded on each lane of a 10% SDS-polyacrylamide gel. The proteins were transferred to nitrocellulose membrane, washed with TBS/0.1% Tween 20, and incubated in 1× TBS-Tween20/2% BSA overnight at 4°C with mouse anti-pAkt(Ser473) antibody (587F11, Cell Signaling Technology, Inc., Danvers, MA) and rabbit anti-pan-Akt (Cell Signaling Technologies, Inc.). The membrane was then washed with TBS/0.1% Tween 20, incubated for 1 h at room temperature with goat anti-mouse 800 nm fluorescent and goat anti-rabbit 680 nm fluorescent labeled antibodies, and scanned on a Li-Cor (Lincoln, NE) Odyssey infrared scanner.
Results
A total of 19 subjects were enrolled on the study between October 15, 2006, and March 19, 2008; six subjects did not receive study drug because of either disease progression prior to treatment or a negative p-AKT result in the study-mandated pretreatment tumor biopsy specimen. Subjects enrolled were heavily pretreated, and because of the requirement that their tumor be accessible for repetitive biopsy, generally had a large tumor burden. Four subjects were enrolled at the 15 mg/m2 dose level, but one experienced disease progression prior to completing the first cycle. The other three all experienced disease progression while on therapy. The most frequently observed toxic effects were gastrointestinal.
Four subjects were enrolled at the 25 mg/m2 dose level; two developed symptomatic and radiographic disease progression, rendering them ineligible to receive study drug. Toxic effects were modest (Table 2) and one subject with metastatic melanoma (cutaneous metastases) received six cycles of therapy before being withdrawn for disease progression. Five subjects were accrued at the 35 mg/m2 dose level; two developed disease progression during their first cycle of therapy and were withdrawn. The other three subjects completed two cycles of therapy before being withdrawn, two because of disease progression, and the third for development of symptomatic syndrome of inappropriate antidiuretic hormone secretion (SIADH), which resolved upon withdrawal of the drug. Five subjects were enrolled at the 45 mg/m2 dose level, but only one received experimental therapy. The remaining four were withdrawn because of disease progression; three had abnormal liver chemistry results due to hepatic progression of disease, rendering them ineligible to receive therapy, and the fourth had interval development of symptomatic brain metastases. The one subject receiving therapy at the 45 mg/m2 dose level had acceptable tolerance of therapy, with no grade 3 or 4 toxic effects observed. In March 2008, the study was terminated due to the primary objective study objective having been met, in addition to the lack of single agent activity observed in this enriched population for activated AKT, and the decreased patient accrual to the study.
Table 2.
Investigator-determined TCN-PM-related toxicity (CTCAE v 3.0)
| Dose level | Number subjects | Toxicity | Grade 1 | Grade 2 | Grade 3 |
|---|---|---|---|---|---|
| 15 mg/m2 | 4 | Anorexia | 1 | 1 | – |
| Nausea | 3 | – | – | ||
| Fatigue | – | – | – | ||
| Sore Tongue | 1 | – | – | ||
| Vomiting | 1 | – | – | ||
| Myalgias | – | 2 | – | ||
| Pruritis | 1 | – | – | ||
| Thrombocytopenia | 1 | – | – | ||
| 25 mg/m2 | 3 | Anorexia | 1 | 2 | – |
| Fatigue | – | 1 | – | ||
| Nausea | 3 | – | – | ||
| Vomiting | 1 | – | – | ||
| Alopecia | 1 | 1 | – | ||
| Myalgia | 1 | – | – | ||
| Neuropathy | 1 | – | – | ||
| 35 mg/m2 | 5 | Prolonged QTc | – | 1 | 1 |
| Anorexia | 1 | 2 | 1 | ||
| Fatigue | 1 | 1 | – | ||
| Nausea | 2 | – | – | ||
| Vomiting | 1 | – | – | ||
| Myalgias | 1 | – | – | ||
| Thrombocytopenia | 1 | – | – | ||
| Hyponatremia | 1 | – | – | ||
| 45 mg/m2 | 1 | Anorexia | – | – | – |
| Nausea | – | 1 | – | ||
| Fatigue | – | – | – |
Nonhematologic toxic effects
The most common investigator-identified toxic effects of TCN-PM were nausea and vomiting, which responded to antiemetic therapy, and fatigue (Table 2). Therapy was well tolerated, and there were only two instances of treatment being withheld because of toxicity. A subject with metastatic soft tissue sarcoma had one episode of grade 3 electrocardiographic QTc prolongation; the patient was not treated further because staging reevaluation confirmed the investigator’s clinical impression of disease progression. The electrocardiographic abnormalities resolved upon withdrawal of the drug. Another patient experienced an episode of grade QTc prolongation in week 4 of cycle 2, which resolved the next week prior to treatment with study drug; this episode did not result in the patient being withdrawn from the study. The episode of SIADH occurred in a subject with retroperitoneal soft tissue sarcoma the during week 3 of cycle 2 of treatment at the 35 mg/m2 dose level; a serum sodium level of 127 mEq/mL, a urine osmolarity of 671 mOsm/kg and a serum osmolarity of 280 mOsm/kg were recorded. Serum sodium levels at study entry and at weekly analysis were normal up to that time; urine and serum osmolarity were not analyzed before treatment. The patient’s serum sodium level recovered to normal upon withdrawal of the study drug, and the subject was withdrawn from the study because imaging studies documented disease progression. Other nonhematologic side effects observed were myalgias, neuropathy and pruritis.
Hematologic toxic effects
One case of grade 1 treatment-related thrombocytopenia was observed; there were no instances of leukopenia. The effect of the drug on erythropoiesis was difficult to assess in this heavily pretreated group of subjects, especially since the use of erythropoietic growth factors was allowed on study; no subjects required red cell transfusion.
Pharmacokinetic and pharmacodynamic evaluation
Plasma samples were obtained from all subjects during their first cycle of treatment for analysis of serum levels of TCN-PM and its metabolite triciribine. The serum concentrations of triciribine and TCN-PM at 2 h and 24 h after infusion are shown in Fig. 2. The pharmacokinetic evaluation of the two compounds was challenging due to a lack of detectable drug levels beyond 24 h in many subjects. This may have been due to the low starting doses. It was anticipated that TCN-PM would produce the metabolite triciribine in vivo producing detectable levels of triciribine in the later sampling. There was also the potential for TCN-PM to re-appear in later sampling. As previously reported, we had findings of unpredictable plasma levels and inter-patient variability [15]. In some subjects, there were occasional instances of a decrease in levels followed by a small increase on the next day, only to follow with undetectable drug the next day. This generally occurred between 24 h to 72 h in the last two dose levels. More importantly, in measurable samples, specifically these 35 mg/m2 and 45 mg/m2 dose levels, there was the appearance of prolonged exposure to both TCN-PM and triciribine well beyond the 24 h time point.
Fig. 2.

Concentration of triciribine (TCN) and TCN-PM following infusion of TCN-PM
Modest decrements in p-AKT levels by IHC were seen at the higher dose levels (35 mg/m2 and 45 mg/m2), although the sample size is too small to draw firm conclusions (Table 3). No significant differences in p-AKT expression were detected by western blot analyses.
Table 3.
Results of tumor P-AKT pre- and during therapy
| Dose level (mg/m2) | Tumor type | Archival P-AKT | Pre-treatment P-AKT | Post-treatment P-AKT |
|---|---|---|---|---|
| 15 | Sarcoma malignant fibrous histiocytoma | 3+ | 2+ | 2+ |
| 15 | Colon cancer | 2+ | 1+ | 1+ |
| 15 | Melanoma | 3+ | 3+ | 3+ |
| 25 | Gastroesophageal cancer | 2+ | 2+ | 2+ |
| 25 | Anal cancer | 3+ | 3+ | No tumor |
| 25 | Sarcoma synovial cell sarcoma | 3+ | 1+ | No tumor |
| 35 | Sarcoma | 3+ | 2+ | 0 |
| 35 | Ovarian cancer | 2+ | 2+/present | 1+/present |
| 35 | Colon cancer | 2+ | 1+ | 0 |
| 45 | Gastroesophageal cancer | 2+ | 2+ | 0 |
Conclusion
AKT plays an important role in cell division and is overexpressed and mutated in a number of malignancies, making it an important target in cancer therapeutics. To our knowledge triciribine is the first small-molecule inhibitor of AKT to be evaluated in a clinical trial in subjects who have tumors with activated AKT, as demonstrated by the presence of p-AKT detected by IHC. Multiple other compounds that inhibit AKT by a variety of mechanisms, both specific and nonspecific, are under evaluation in both preclinical and human subject testing. Antisense therapy specific to AKT1 has been developed (RX-0201, Rexahn Corporation) that, when administered by continuous infusion, has an MTD of 315 mg/m2/day; the DLT was fatigue [17]. The orally available synthetic alkylphospholipid perifosine has been demonstrated to inhibit AKT phosphorylation in vitro. Phase I trials have been completed, and it has been evaluated as single-agent therapy in phase II trials in advanced soft tissue sarcoma, head and neck cancer, prostate cancer, pancreatic cancer and Waldenström’s macroglobulinemia [18–22]. Radiographic responses were observed in the solid tumor studies; a 35% response rate (3% partial and 32% minimal based on serum immunoglobin levels was reported) [22]. No prior phase I or phase II study of an inhibitor of p-AKT included tumor tissue target validation as an objective of the trial.
Many new ATP-competitive and allosteric AKT kinase inhibitors have been developed commercially; several of these drugs have entered clinical testing, both as single agents and in combination with cytotoxic chemotherapy agents [23]. The desirability of selectively inhibiting specific isoforms of AKT compared to inhibiting all three isoforms is unknown at this time.
Although inhibiting the PI3K/AKT pathway would appear a promising new strategy for cancer therapeutics, some preclinical data suggest that AKT has antimetastatic activity. Preclinical data have shown that activated AKT inhibits breast cancer cell motility and invasion by inhibiting the transcriptional factor NFAT [24]. In a transgenic mouse model of tumors that overexpress HER2/neu, primary tumors associated with high levels of p-AKT were larger, but their host mice had fewer metastases [25]. These data suggest that AKT inhibition in some settings may lead to unintended effects, such as stimulation of metastasis; for this reason, therapy with AKT inhibitors may be deployed clinically most effectively in combination with other cell cycle- or pathway-specific inhibitors [26].
Insufficient tumor tissue remained after p-AKT analysis by IHC and WB to evaluate tumor adenosine kinase levels. TCN-P is a prodrug of triciribine and is dephosphorylated in the serum to triciribine, which can cross the cell membrane; within the cell, triciribine requires phosphorylation by adenosine kinase at the 5′ position to exert its anticancer activity. Previous in vitro studies of 60 cell lines showed a relationship between tumor adenosine kinase levels and the activity of TCN-P (as measured by the drug’s ability to inhibit growth by 50%, GI50) [27]. It is possible that the lack of clinical activity in this trial might reflect tumor selection, in that a disproportionate number of the tumors may have had low levels of tumor adenosine kinase.
While published trials had demonstrated that this drug had radiographic activity in the treatment of cervical cancer [10], no significant radiographic responses were seen in this group of patients selected for tumor p-AKT expression. Although none of the subjects appeared to receive significant clinical benefit from the experimental agent, there did not appear to be acceleration of their disease while on therapy. The predetermined primary objective of the study was to evaluate whether suppression of AKT phosphorylation could be demonstrated by analysis of tumor samples prior to, and following, therapy with TCN-PM; modest decreases in p-AKT were demonstrated in tumor samples taken 24 h following the second dose of TCN-PM (cycle 1, day 15 dose). Although the number of patients studied was small, this finding supports the hypothesis that in vitro inhibition of AKT phosphorylation can be effected by treatment with TCN-PM. While the study was terminated early due to a number of factors, including response to the lack of potential benefit to research subjects, future studies with TCN-PM, both as monotherapy (in hematologic malignancies) and combination therapy (with other cytosolic pathway inhibitors and cytotoxic agents should evaluate tumor p-AKT levels prior to and following treatment with this agent, to improve our understanding of this agent’s mechanisms of action. It is not clear at this time as to whether AKT phosphorylation as measured by IHC is a predictive biomarker of response for AKT phopshorylation inhibitors, and future biomarkers may be identified which predict patients most likely to benefit from these class of agents.
Acknowledgments
Grant Support: Vioquest Pharmaceuticals, CA107078 and 1KG02-33967.
We would like to thank Katherine Stemke Hale, Shrikanth Reddy and Milind Javle for their thoughful review of the manuscript.
Footnotes
Preliminary data from this study were presented at the 16th AACR-NCI-EORTC: Molecular Targets and Cancer Therapeutics Symposium, 2007, San Francisco, California.
Financial disclosures Dr. Said Sebti is a consultant for Vioquest Pharmaceticals.
Contributor Information
Christopher R. Garrett, Department of Experimental Therapeutics, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA. Department of Oncologic Sciences, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA
Domenico Coppola, Department of Experimental Therapeutics, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA. Department of Oncologic Sciences, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA.
Robert M. Wenham, Department of Experimental Therapeutics, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA. Department of Oncologic Sciences, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA
Christopher L. Cubitt, Department of Experimental Therapeutics, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA. Department of Oncologic Sciences, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA
Anthony M. Neuger, Department of Experimental Therapeutics, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA. Department of Oncologic Sciences, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA
Timothy J. Frost, Department of Experimental Therapeutics, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA. Department of Oncologic Sciences, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA
Richard M. Lush, Department of Experimental Therapeutics, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA. Department of Oncologic Sciences, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA
Daniel M. Sullivan, Department of Experimental Therapeutics, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA. Department of Oncologic Sciences, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA
Jin Q. Cheng, Department of Molecular Oncology, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA. Department of Oncologic Sciences, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA
Saïd M. Sebti, Department of Drug Discovery, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA. Department of Oncologic Sciences, H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa, FL 33612-9497, USA
References
- 1.Bellacosa A, Testa JR, Staal SP, et al. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 2.Bellacosa A, de Feo D, Godwin AK, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280–285. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- 3.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 4.Sun M, Wang G, Paciga JE, et al. AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol. 2001;159:431–437. doi: 10.1016/s0002-9440(10)61714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng JQ, Altomare DA, Klein MA, et al. Transforming activity and mitosis-related expression of the AKT2 oncogene: evidence suggesting a link between cell cycle regulation and oncogenesis. Oncogene. 1997;12:2793–2801. doi: 10.1038/sj.onc.1201121. [DOI] [PubMed] [Google Scholar]
- 6.Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 7.Schram KH, Townsend LB. The synthesis of 6-amino-4-methyl-8-(β-D-ribofuranosyl (4-H)pyrrolo-[4-3-2depyrimido(4, 5-C) pyridazine, a new tricyclic nucleoside. Tetrahedron Lett. 1971;49:4757–4760. [Google Scholar]
- 8.Feun LG, Savaraj N, Bodey GP, et al. Phase I study of tricyclic nucleoside phosphate using a five-day continuous infusion schedule. Cancer Res. 1984;44:3608–3612. [PubMed] [Google Scholar]
- 9.Mittelman A, Casper ES, Godwin TA, et al. Phase I study of tricyclic nucleoside phosphate. Cancer Treat Rep. 1983;67:159–162. [PubMed] [Google Scholar]
- 10.Schilcher RB, Haas CD, Samson MK, et al. Phase I evaluation and clinical pharmacology of tricyclic nucleoside 5′-phosphate using a weekly intravenous regimen. Cancer Res. 1986;46:3147–3151. [PubMed] [Google Scholar]
- 11.Feun LG, Blessing JA, Barrett RJ, et al. A phase II trial of tricyclic nucleoside phosphate in patients with advanced squamous cell carcinoma of the cervix. A Gynecologic Oncology Group Study. Am J Clin Oncol. 1993;16:506–508. doi: 10.1097/00000421-199312000-00010. [DOI] [PubMed] [Google Scholar]
- 12.Hoffman K, Holmes FA, Fraschini G, et al. Phase I–II study: triciribine (tricyclic nucleoside phosphate) for metastatic breast cancer. Cancer Chemother Pharmacol. 1996;37:254–258. doi: 10.1007/BF00688325. [DOI] [PubMed] [Google Scholar]
- 13.O’Connell MJ, Rubin J, Hahn RG, et al. Phase II clinical trial of tricyclic nucleoside phosphate for advanced colorectal cancer. Cancer Treat Rep. 1987;71:333–334. [PubMed] [Google Scholar]
- 14.Yang L, Dan HC, Sun M, et al. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64:4394–4399. doi: 10.1158/0008-5472.CAN-04-0343. [DOI] [PubMed] [Google Scholar]
- 15.Powis G, Basseches PJ, Kroschel DM, et al. Disposition of tricyclic nucleoside-5′-monophosphate in blood and plasma of patients during phase I and II clinical trials. Cancer Treat Rep. 1986;70:359–362. [PubMed] [Google Scholar]
- 16.Cancer Therapy Evaluation Program. Common terminology criteria for adverse events, version 3.0. Department of Health and Human Services, National Institutes of Health, National Cancer Institute; 2003. Mar 31, [Google Scholar]
- 17.Marshall J, Posey J, Hwang J, et al. A phase I trial of RX-0201 (AKT anti-sense) in patients with an advanced cancer. Proc Am Soc Clin Oncol. 2007:abstr 3564. [Google Scholar]
- 18.Knowling M, Blackstein M, Tozer R, et al. A phase II study of perifosine (D-21226) in patients with previously untreated metastatic or locally advanced soft tissue sarcoma: a National Cancer Institute of Canada Clinical Trials Group trial. Invest New Drugs. 2006;24:435–439. doi: 10.1007/s10637-006-6406-7. [DOI] [PubMed] [Google Scholar]
- 19.Argiris A, Cohen E, Karrison T, et al. A phase II trial of perifosine, an oral alkylphospholipid, in recurrent or metastatic head and neck cancer. Cancer Biol Ther. 2006;5:766–770. doi: 10.4161/cbt.5.7.2874. [DOI] [PubMed] [Google Scholar]
- 20.Chee KG, Longmate J, Quinn DI, et al. The AKT inhibitor perifosine in biochemically recurrent prostate cancer: a phase II California/Pittsburgh cancer consortium trial. Clin Genitourin Cancer. 2007;5:433–437. doi: 10.3816/CGC.2007.n.031. [DOI] [PubMed] [Google Scholar]
- 21.Hedley D, Moore MJ, Hirte H, et al. A Phase II trial of perifosine as second line therapy for advanced pancreatic cancer. A study of the Princess Margaret Hospital Phase II Consortium. Proc Am Soc Clin Oncol. 2005:abstr 4166. [Google Scholar]
- 22.Ghobrial IM, Leleu X, Rubin N, et al. Phase II trial of the novel oral Akt inhibitor perifosine in relapsed and/or refractory Waldenström macroglobulinemia. Proc Am Soc Clin Oncol. 2008:abstr 8546. [Google Scholar]
- 23.Lindsley CW, Barnett SF, Layton ME, et al. The PI3K/Akt pathway: recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr Cancer Drug Targets. 2008;8:7–18. doi: 10.2174/156800908783497096. [DOI] [PubMed] [Google Scholar]
- 24.Yoeli-Lerner M, Yiu GK, Rabinovitz I, et al. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol Cell. 2005;20:539–550. doi: 10.1016/j.molcel.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 25.Hutchinson JN, Jin J, Cardiff RD, et al. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppress tumor invasion. Cancer Res. 2004;64:3171–3178. doi: 10.1158/0008-5472.can-03-3465. [DOI] [PubMed] [Google Scholar]
- 26.Sawyers CL. Will kinase inhibitors have a dark side? N Engl J Med. 2006;20:313–315. doi: 10.1056/NEJMcibr062354. [DOI] [PubMed] [Google Scholar]
- 27.Shedden K, Townsend LB, Drach JC, et al. A rational approach to personalized anticancer therapy: chemoinformatic analysis reveals mechanistic gene-drug associations. Pharm Res. 2003;20:843–847. doi: 10.1023/a:1023893700386. [DOI] [PubMed] [Google Scholar]