

Abstract
Lysine 2,3-aminomutase (LAM) is a radical S-adenosyl-L-methionine (SAM) enzyme and, like other members of this superfamily, LAM utilizes radical-generating machinery comprising SAM anchored to the unique Fe of a [4Fe-4S] cluster via a classical five-membered N,O chelate ring. Catalysis is initiated by reductive cleavage of the SAM S–C5′ bond, which creates the highly reactive 5′-deoxyadenosyl radical (5′-dAdo•), the same radical generated by homolytic Co–C bond cleavage in B12 radical enzymes. The SAM surrogate S-3′,4′-anhydroadenosyl-L-methionine (anSAM) can replace SAM as a cofactor in the isomerization of L-α-lysine to L-β-lysine by LAM, via the stable allylic anhydroadenosyl radical (anAdo•). Here electron nuclear double resonance (ENDOR) spectroscopy of the anAdo• radical in the presence of 13C, 2H, and 15N-labeled lysine completes the picture of how the active site of LAM from Clostridium subterminale SB4 “tames” the 5′-dAdo• radical, preventing it from carrying out harmful side reactions: this “free radical” in LAM is never free. The low steric demands of the radical-generating [4Fe-4S]/SAM construct allow the substrate target to bind adjacent to the S–C5′ bond, thereby enabling the 5′-dAdo• radical created by cleavage of this bond to react with its partners by undergoing small motions, ~0.6 Å toward the target and ~1.5 Å overall, that are controlled by tight van der Waals contact with its partners. We suggest that the accessibility to substrate and ready control of the reactive C5′ radical, with “van der Waals control” of small motions throughout the catalytic cycle, is common within the radical SAM enzyme superfamily and is a major reason why these enzymes are the preferred means of initiating radical reactions in nature.
INTRODUCTION
Lysine 2,3-aminomutase (LAM) is a member of the radical S-adenosyl-L-methionine (SAM) enzyme superfamily,1,2 whose reactions are initiated by radical-generating machinery comprising SAM anchored to the unique Fe of a [4Fe-4S] cluster via a classical five-membered N,O chelate ring formed by the methionine.3 This novel structural motif for radical generation was discovered by electron nuclear double resonance (ENDOR) spectroscopy of pyruvate formatelyase activating enzyme (PFL-AE)4–6 and LAM,7 and subsequently visualized by X-ray structure determination of these two enzymes as well as other superfamily members.1,8–10 Electron transfer from the [4Fe4S]1+ cluster initiates radical SAM reactions by reductive cleavage of the S–C5′ bond to create the highly reactive 5′-deoxyadenosyl radical (5′-dAdo•) (Scheme 1).1
Scheme 1.
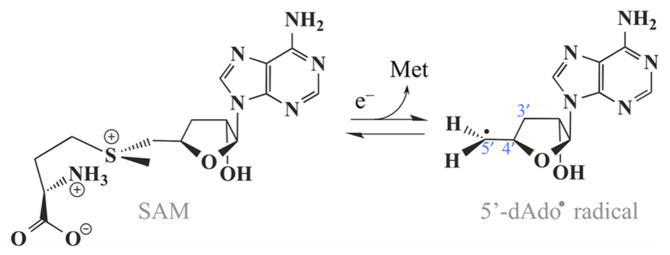
The 5′-dAdo• radical is common to both radical SAM and B12-dependent enzymes, with the latter generating this radical through homolytic cleavage of the Co–C5′ bond of the B12 coenzyme.11
LAM utilizes the radical-SAM machinery to isomerize L-α-lysine to L-β-lysine (Scheme 2) in a reaction directly analogous to the B12-dependent isomerases.11–13
Scheme 2.
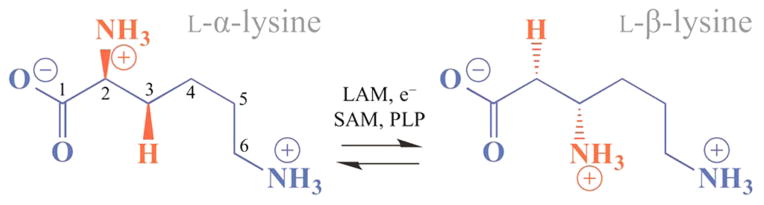
The reactive 5′-dAdo• radical formed by reductive cleavage of SAM (Figure 1, state 2)1 abstracts an H atom from substrate bound to pyridoxal 5′-phosphate (PLP) to form 5′-deoxyade-nosine (5′-dAdoH) and the α-Lys• radical (Lys•) (state 3). This radical isomerizes to the β-Lys• radical (state 4), which then abstracts an H atom from 5′-dAdoH to form β-Lys and 5′-dAdo• radical (state 5); the latter then regenerates SAM. A schematic representation of these states is provided in Figure 1, with individual states numbered for easy reference throughout this paper.
Figure 1.
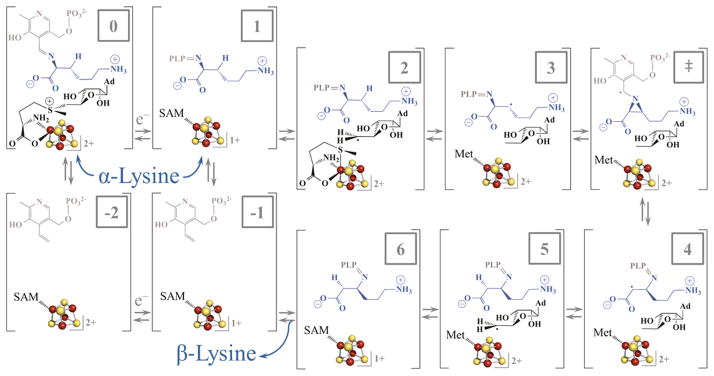
Steps in the reaction catalyzed by LAM. State 0 is the oxidized [4Fe-4S]2+ state with SAM coordinated to the unique iron of the cluster and substrate Lys present as a Schiff base. One-electron reduction of the cluster provides state 1, which initiates reductive cleavage of SAM to generate the 5′-dAdo• in state 2. States 3–6 represent the radical rearrangement of α-lysine to β-lysine, abstraction of H• from 5′-dAdoH, and reformation of SAM. PLP is depicted in grey, while lysine is in blue.
Multinuclear ENDOR spectroscopy (13C, 1,2H, 31P, and 14N) led us to propose that inner sphere electron transfer from the reduced [4Fe-4S]+ cluster cleaves the S–C5′ bond of SAM to form the 5′-dAdo• radical, while the sulfur of methionine becomes the sixth ligand to the unique iron.7 This approach also was used to characterize the active site of LAM in intermediate states that contain the isomeric substrate radicals or analogues.14 With L-α-lysine as substrate, the β-Lys• radical (state 4) produced by H atom abstraction and rearrangement was monitored. In parallel, state 3 was probed through use of two substrate analogues (trans-4,5-dehydro-L-lysine and 4-thia-L-lysine), which generate stable analogues of the α-Lys• radical. This study provided a first glimpse of the motions of active site components during catalytic turnover, and suggested a possible major movement of PLP during catalysis. The principal focus of that work, however, was on the relative positions of the carbons involved in H atom transfer. By use of ENDOR to study hyperfine couplings to the substrate radicals it was concluded that the active site facilitates hydrogen atom transfer by enforcing van der Waals contact between radicals and their reacting partners, and that this constraint “tames” the highly reactive 5′-dAdo• radical, enabling the enzyme to minimize and even eliminate side reactions of this highly reactive species.
Noticeably absent from this or any study of radical SAM enzymes was any direct examination of the central event in the catalytic cycle of a radical SAM enzyme: the creation of the 5′-dAdo• radical by reductive S–C5′ bond cleavage, and its subsequent migration to the site of H atom abstraction from substrate (state 2). Of all steps in the cycle, one would presume this must be the most tightly controlled, so as to occur without side reaction of the 5′-dAdo• radical with the environment. The intermediate 5′-dAdo• radical, however, is so reactive that it has never been observed, either in radical SAM or B12-dependent enzymes, precluding characterization of this step.
To overcome this obstacle, Frey, Reed, and co-workers developed the SAM surrogate, S-3′,4′-anhydroadenosyl-L-methionine (anSAM).15,16 The compound is a true cofactor for LAM, catalyzing the isomerization shown in Scheme 2 with a specific activity of 0.10 ± 0.02 IU/mg, compared to 35–40 IU/mg for the natural cofactor SAM.15 However, when the [4Fe-4S]1+ donates an electron to anSAM, cleaving the S–C5′ bond, this forms the stable allylic anhydroadenosyl radical (anAdo•) (Scheme 3) which can be used to investigate active site rearrangements through use of ENDOR spectroscopy. Once the catalytically competent 5′-dAdo• or anAdo• radicals are “born”, the active site must than precisely shepherd the radical to the substrate site of H atom abstraction, so as to avoid side reactions, a process that has been incisively studied with B12-dependent enzymes.17–25 We here probe this rearrangement in LAM through the use of 13C/1,2H/15N ENDOR to monitor the distances and interactions between the anAdo• radical and multiple sites of isotopically labeled Lys substrate, especially the site of H atom abstraction. We also probe the distance to the –S13CH3 of the methionine formed by SAM cleavage.
Scheme 3.
Comparison of these ENDOR results for state 2 with the X-ray crystal structure of state 0, both of enzyme from C. subterminale, reveals how the active site of LAM “tames” the 5′-dAdo• radical: this “free radical” is never free. Substrate binds with the 3-C atom of Lys, from which the target H is abstracted, adjacent to the S–C5′ bond of SAM, Figure 2(A), an arrangement similar to that found in all other structures of radical SAM enzymes with bound substrate.10 As a result, upon cleavage of the S–C5′ bond only minimal motions of the 5′-dAdo• radical are needed for it to carry out H atom abstraction from substrate, during which the radical is chaperoned by van der Waals contact with its reaction partners. Integration of this study with our previous study14 of states 3 and 4 completes a picture in which side reactions of the 5′-dAdo• radical are eliminated both at the beginning and the end of the catalytic cycle because this radical undergoes only small motions, which are controlled by “van der Waals constraints”.
Figure 2.
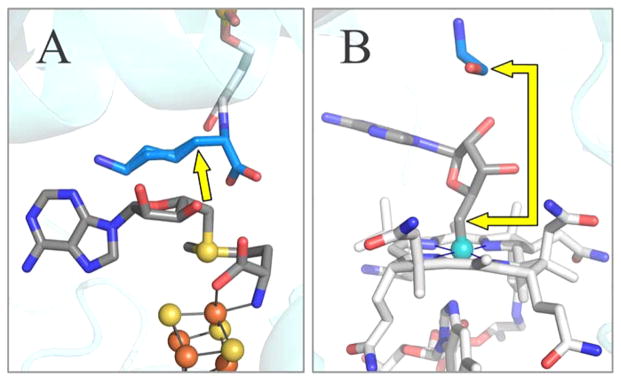
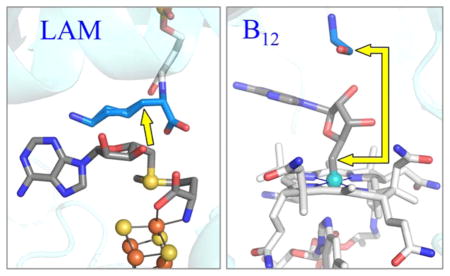
Active site structure of LAM (A) and ethanolamine ammonia-lyase (EAL) (B). The illustration of EAL contains the B12 cofactor as modeled from the crystal structure of EAL with the analogue adeninylpentylcobalamin. The yellow arrows show the distance from C5′ prior to S–C5′ or Co–C5′ bond cleavage, to target carbon atom in the substrate. PDB IDs are 2A5H (LAM) and 3ABS (ethanolamine ammonia-lyase). Carbon atoms are colored as: blue for substrate, dark grey for adenosine and SAM, and light grey for cobalamin and PLP.
We suggest that the accessibility of the C5′ radical to highly diverse substrates is a major reason why evolution spread the use of this construct across all of the kingdoms of life. In contrast, for B12 enzymes, the steric demands of the cofactor force the substrate target to bind farther from the nascent radical at C5′, Figure 2(B), and C5′ must therefore migrate larger distances to reach the target.17–23 The high steric constraints of the coenzyme B12 thus may explain why evolution has found so few uses for this radical generating machinery, which is limited to a few reactions involving small molecules, and to the animal kingdom.11,13,26–30
MATERIALS AND METHODS
Materials
Adenosine triphosphate (ATP), L-methionine, L-cysteine, L-lysine, pyridoxal 5′-phosphate (PLP), adenylate kinase, phosphocreatine kinase, and phosphocreatine were purchased from Sigma. Uniformly labeled 13C and 15N L-lysine were purchased from Cambridge Isotope Laboratories. D,L-lysine-1,2-13C and D,L-lysine-2-13C were purchased from Aldrich. L-Lysine-2,6,6-2H, L-lysine-3,3,4,4,5,5,6,6-2H and D,L-lysine-2-15N were purchased from CDN Isotopes. Sodium dithionite was purchased from Acros Organics and ammonium iron(II) sulfate was purchased from Fisher Chemicals. L-[Methyl-13C]-methionine was purchased from Cambridge Isotope Laboratories. All labeled substrates had a minimum of 99 atom % labeling. L-[U-14C] lysine was obtained from NEN Life Sciences Products. SAM and anSAM were prepared in our laboratory as described in the following sections. All other chemicals and reagents were of the highest purity and used as supplied.
Preparation of LAM
All experiments described herein utilized LAM from Clostridium subterminale SB4. Genetic constructs containing the LAM gene from C. subterminale SB4 (a gift from Perry Frey) were appended with a C-terminal 6-histidine tag by ligation into pET-23a and then transformed into Escherichia coli BL21(DE3)pLysS (Stratagene) cells for protein overexpression. The transformation was plated on LB agar plates containing 50 μg/mL of ampicillin. A single colony was used to inoculate 50 mL of LB media containing ampicillin in a 37 °C incubator shaker at 250 rpm. The overnight growth was then transferred to a New Brunswick Bioflo 110 anaerobic fermentor (10 L) with nutrient enriched LB/ampicillin, and grown with shaking at 250 rpm to an optical density (OD) of ~0.5 at 600 nm. Once the desired OD was reached, protein expression was induced with IPTG (1 mM final concentration) and supplemented with ferrous ammonium sulfate and zinc sulfate. Cells were then allowed to grow with shaking for an additional 2 h, then they were cooled to approximately 30 °C and additional ferrous ammonium sulfate was added. The culture was ultimately cooled to 4 °C and purged with nitrogen for an additional 15 h. The cells were then centrifuged and the resulting cell pellets were stored at −80 °C until further use.
Cell lysis and protein purification were carried out under anaerobic conditions in a Coy chamber. Cell pellets were thawed and resuspended in a lysis buffer containing 50 mM EPPS, 300 mM KCl, 0.1 mM lysine, 10 μM PLP, pH 8.0, 1% Triton X-100, 10 mg PMSF, 9 mg lysozyme, ~1 mg RNase A and DNase I per 50 mL, respectively. The lysis mixture was stirred for ~1 h and then the lysate was centrifuged in gastight bottles at 18 000 rpm for 30 min. The resulting supernatant was loaded onto a HisTrap Ni2+ affinity column (GE healthcare) equilibrated with 50 mM EPPS, 300 mM KCl, 0.1 mM L-lysine, 10 μM PLP, 10 mM imidazole, pH 8.0 (buffer A). Buffer A was utilized for column equilibration and washing. The protein elution was accomplished by increasing the imidazole concentration in a stepwise manner from 10 to 20 to 50 to 75 to 100% using buffer B (50 mM EPPS, 300 mM KCl, 0.1 mM L-lysine, 10 μM PLP, pH 8.0, 500 mM imidazole). Eluted protein (20 and 50% B) was concentrated using a Millipore (13–14 kDa MWCO) spin concentrator. After purification and concentration, the protein was flash frozen in liquid N2 and stored at −80 °C or in liquid N2 until further use. The concentrated protein was dialyzed in 50 mM EPPS, 1 mM dithiothrietol, 0.1 mM L-lysine, 10 μM PLP, pH 8.0 prior to reconstitution. The concentration of the protein was determined by the Bradford31 method, and the iron analysis was done by the colorimetric iron assay described by Fish.32
A reconstitution was performed after protein centrifugation and incubation with dithiothreitol (5 mM) similar to that previously described.33 Sodium sulfide nonahydrate and ferrous ammonium sulfate were added incrementally over 1–2 h, with gentle stirring, to a final concentration of 6× the protein concentration. The solution was incubated for 6 h at 4 °C before being centrifuged, concentrated and passed over a desalting G25 column in buffer (50 mM EPPS, 1 mM DTT, 10 μM PLP, pH 8.0). The protein sample was then concentrated further. All purification was performed in an anaerobic coy chamber.
Preparation of anSAM
The synthesis of (3′,4′)-anhydroadenosine and subsequent preparation of anSAM was similar to that described by Magnusson and Frey,16 but with alterations as described herein. The enzymatic reaction implemented to phosphorylate (3′,)-anhydroadenosine requires adenylate kinase, adenosine kinase, and phosphocreatine kinase. The adenylate kinase and the phosphocreatine kinase were obtained from commercial sources and dialyzed into the assay buffer. The adenosine kinase was prepared in our lab with methods similar to those described previously,34 with the following adjustments. Human T cell cDNA (Molt 14) was a generous gift of the Jutila lab at MSU, and was used for amplification of the adenosine kinase gene with the following promoters:
Forward: 5′ CGG GCA GCA TAT GAC GTC AGT CAG AGA AA 3′.
Reverse: 5′ GCG TTC GGA TCC ATC AGT GGA AGT CTG G 3′.
The amplified DNA was ligated into the T7 based expression vector, pET-14b (Novagen), which also introduces a polyhistidine tag for protein purification. The plasmid with insert was transformed into XL1 Blue supercompetent cells for plasmid amplification, and the purified plasmid was then transformed into BL21(DE3)pLysS for protein expression. Successful expression of this protein required culture temperatures near 22 °C due to formation of inclusion bodies at higher temperatures. Protein expression was induced by addition of IPTG at OD600nm of approximately 0.5; the growth was supplemented with glucose (2 mg/mL) ~5 h after induction and harvested 22 h after induction. The cells were lysed in Tris pH 7.5 buffer (10 mL/g of cells) containing lysozyme (0.2 mg/mL), PMSF (0.5 mg/mL), DNase I (10 μg/mL), RNase A (10 μg/mL), MgSO4 (1 mM), and dithiothreitol (1 mM), by sequentially freezing and thawing the suspension three times. The clarified lysate was fractionated by ammonium sulfate precipitation, and the adenosine kinase precipitated in the 65% to 100% ammonium sulfate cut. The precipitated adenosine kinase was dissolved in 10 mM Tris-HCl/1 mM DTT pH 7.5 and dialyzed against the same buffer before applying it to a HisTrap column equilibrated with 20 mM Tris-HCl/100 mM KCl/10 mM imidazole (pH 7.5). The protein was eluted using a gradient to 20 mM Tris-HCl/100 mM KCl/500 mM imidazole (pH 7.5), eluting at ~182 mM imidazole. After buffer exchange into Tris-HCl (10 mM, pH 7.5) with dithiothreitol (1 mM), the AdoK was loaded onto an HR16/10 DEAE Sephacel column equilibrated with the same buffer. AdoK was eluted with a linear gradient of 0–100 mM KCl in the same buffer, eluting at ~40 mM KCl. The AdoK was then exchanged into PIPES (25 mM), MgCl2 (1.25 mM), pH 7.5.
In order to convert anhydroadenosine to anhydroATP, PIPES (25 mM), MgCl2 (1.25 mM), BSA (0.01% w/v), anhydroadenosine (3 mM), phosphocreatine (20 mM), adenosine kinase (1000 IU), adenylate kinase (2000 IU), and phosphocreatine kinase (2000 IU) were combined while maintaining pH at approximately 7.0; GTP (0.6 mM) was added to initiate the phosphorylation. The reaction was monitored by TLC and quantified by HPLC. Upon completion of the phosphorylation reactions, the anhydroadenosine triphosphate (anATP) was purified using a HR10/30 DEAE Sephadex A25 column with a linear gradient of NH4HCO3 (10–500 mM); the anATP elutes around 350 mM ammonium bicarbonate. The anATP was then rotovapped or lyophilized to a solid.
SAM synthetase was expressed and purified as described by Markham et al.,17 with some minor modifications.4 In a reaction similar to that described by Park et al.35 and modified by us previously4 and further modified herein, we utilized SAM synthetase to enzymatically transform anhydroadenosine triphosphate into anhydro-S-adenosyl-L-methionine (anSAM) in EPPS buffer (50 mM, pH 8.0). The anSAM was then purified on a Source 15S cation exchange column with a linear gradient of 0 to 1 M HCl, as confirmed by NMR (Bruker Cryomagnet BZH 500/51; 64.2A, 11.70 T) and LC–MS (Agilent HPLC 1100 with Bruker MicroTOF). AnSAM containing 13C in the methyl group was synthesized from ATP and L-[methyl-13C]-methionine by use of SAM synthetase-catalyzed reactions as described herein.4
Preparation of ENDOR Samples
ENDOR samples were prepared under anaerobic conditions in a Coy anaerobic chamber. The purified LAM (0.90 mM LAM, ~270 μM active sites) was activated by incubation with 10 mM L-cysteine in the presence of 0.90 mM PLP, 1.0 mM ammonium iron(II) sulfate, and 15% glycerol in 42 mM EPPS buffer at pH 8.0 and 37 °C for 4 h. The activated enzyme was concentrated with Microcon 30 centrifugal filter devices (Millipore) so that the concentration of enzyme active site was ~1.2 mM. The concentrated enzyme was quickly mixed with anSAM (or [methyl-13C]-anSAM), sodium dithionite, and L-lysine (one of the following: unlabeled L-lysine, [u-13C,15N]-L-lysine, [1,2-13C]-D,L-lysine, [2-13C]-D,L-lysine, [2,6,6-2H]-L-lysine, [3,3,4,4,5,5,6,6-2H]-L-lysine, [2-15N]-D,L-lysine) in a 0.65 mL microcentrifuge tube. The mixed sample was rapidly transferred to an ENDOR sample tube and frozen in liquid nitrogen. The time scale from mixing to sample freezing was from 1 to 1.5 min, sufficient time for equilibrium to be attained in the reaction of 0.9 mM LAM. In all samples, the concentrations of the enzyme active site were ~0.9 mM; the concentrations of anSAM, sodium dithionite, and lysine (or its analogue) were 3.0, 6.0, and 40 mM, respectively. All assays were performed in an MBraun anaerobic chamber at <1 ppm oxygen.
EPR and ENDOR Spectroscopy
X-band CW electron paramagnetic resonance (EPR) measurements were performed by a Bruker ESP 300 spectrometer equipped with an Oxford Instruments ESR 910 continuous He flow cryostat.
Q-band CW ENDOR spectra were collected on a spectrometer with a helium immersion dewar previously reported.36,37 All measurements were done at 2 K. Stochastic field-modulation detected ENDOR technique, which has been first reported by Brueggeman and Niklas,38 was here utilized for 1H ENDOR measurement. In the stochastic ENDOR sequence, RF is randomly hopped over the range of spectrum region with the subtraction of background signal (RF off) at each frequency.
Pulsed ENDOR spectra were collected on a spectrometer described earlier,19 equipped with a helium immersion dewar for measurements at 2 K. ENDOR measurements employed the Mims pulse sequence (π/2–τ–π/2–T–π/2–τ–echo, RF applied during interval T) for small hyperfine couplings or the Davies pulse sequence (π–T–π/2–τ–π–τ–echo) for large hyperfine couplings.39 To determine the sign of hyperfine couplings we utilized the recently developed Pulsed ENDOR Saturation and Recovery (PESTRE) technique, which employs a multipulse sequence comprised of the Davies ENDOR sequence.40
For nuclei (N) of spin I = 1/2(13C, 1H, 15N) interacting with a S = 1/2 paramagnetic center, the first-order ENDOR spectrum for a single molecular orientation is a doublet with frequencies (ν+/ν−),
| (1) |
where νN is the Larmor frequency and A is the orientation-dependent hyperfine constant. For nuclei with I = 1 (2H), the first-order ENDOR condition can be written:
| (2) |
where P is the orientation-dependent quadrupolar splitting.
For a nucleus with hyperfine coupling, A, Mims pulsed ENDOR has a response R that depends on the product, Aτ, according to the equation,
| (3) |
This function has zeros, corresponding to minima in the ENDOR response (hyperfine “suppression holes”), at Aτ = n; n = 0, 1, …, and maxima at Aτ = (2n + 1)/2; n = 0, 1, …8,20 The “holes” at A = n/τ, n = 1, 2, 3, …, can be adjusted by varying τ. However, the “central”, n = 0, hole at ν = νN persists regardless. This can be of significance in distinguishing a tensor that is dominated by anisotropic interactions from one that is dominated by isotropic ones. The latter would never lead to ENDOR intensity near νN; the former does so for certain orientations, but the ν = 0 Mims hole tends to diminish the differences between the two cases.
Neither the very small dispersion of g values nor the hyperfine coupling to the nuclei of anAdo• radical are resolved in the 35 GHz EPR spectra of state 2 of LAM prepared with anSAM. Consequently, ENDOR spectra of nuclei hyperfine-coupled to the radical collected at the field of maximum EPR intensity are an isotropic powder-averaged pattern. Simulation of these powder patterns for nuclei of the anAdo• radical itself are analyzed in terms of the spin distribution among the carbons of the allyl fragment as a test of the reported spin distribution.16 Simulation of the ENDOR patterns for nuclei of substrate yield hyperfine tensors that allow estimation of distances between the radical center and those nuclei, as well as bonding and orbital overlap properties, but do not yield orientations relative to the g-frame or molecular frame. All simulations were done with the program, ENDORSIM.41
The central goal of these measurements is to determine the position of the reactive C5′ carbon relative to the atoms of the L-Lys substrate through analysis of the hyperfine coupling tensor, A, between the nuclei of Lys and C5′ of the anAdo• radical. Such a tensor is the sum of a possible contribution from spin delocalization across the noncovalent “interface” between anAdo• radical and atoms of Lys (see Discussion), the so-called isotropic coupling (coupling constant, Aiso), and the through-space dipolar coupling between the nucleus of Lys and the spin of the anAdo• radical. The distance-dependence of this latter term yields the sought-for metrical result.
In principle, the dipolar term is the sum of the interactions with the spin density on all three allyl carbons, the two with large spin densities ρ(C5′) ~ ρ (C3′) ~ 0.59, and the one with small spin density, ρ (C4′) ~ −0.18.16 However, C5′ of anAdo• is close to or even in van der Waals contact with Lys nuclei, whereas the other carbon with high spin density, C3′, is much farther away, and C4′ has low spin density, and so we began our determination of distances between C5′ and atoms of Lys by ignoring the contribution from spin on the allyl carbons other than C5′. In this case the through-space hyperfine coupling to anAdo• radical spin on C5′ is described by the axial form
| (4) |
In the point-dipole approximation for the electron spin, the parameter T is related to the distance from C5′ to the nucleus being examined through the relationship,
| (5) |
where ρ(C5′) = +0.59. As described in the Discussion, at the (relatively) short distances associated with 2-13C and 3-2H: one must take into account a correction to the point-dipole equations associated with the distributed nature of the spin in the 2pπ orbital of C5′ through use of equations presented by McConnell and Strathdee.42
We tested the validity of the ignoring the effects of spin density on C4′ and C3′ in the determination of distances between C5′ and atoms of Lys, in the following fashion. As described below we used the distances between C5′ and various nuclei of Lys as calculated through the use of spin density on C5′ alone to estimate the position of C5′ of anAdo• radical, then approximated the position of the remainder of the radical by simply translating the Ado fragment present in the state 5 crystal structure so as to place C5′ at that position (see Figure 7, below). The positions of C4′ and C3′ within this heuristic structure were then used as input into previously published equations43 to test whether the spin density on those atoms materially alters the dipolar interaction tensor, T, and hence the calculated distances of C5′ to the Lys nuclei. The extended treatment shows that the small negative spin density on C4′ adjacent to C5′, ρ(C4′) = −0.18, would tend to slightly shorten the estimated distance from C5′ to target atoms and to introduce a small rhombicity (|T2| > |T1|, eq 4) to the coupling tensor. However, within the precision of these calculations, this effect is countered by the effect of the large positive spin density, ρ(C3′) = +0.59, on the more remote C3′ to lengthen the calculated distance and to introduce a rhombicity of the opposite sense (|T2| < |T1|, eq 4), the two effects thus largely cancel each other; most importantly even when the extended model predicted a non-negligible rhombicity, it gave a value of 2T, the largest dipolar splitting, not significantly different from that obtained by using the distance to C5′ in eq 5. Hence, for the purposes of this paper, the simplified analysis in terms of spin density on C5′ alone is satisfactory.
Figure 7.

Inferred motion of 5′-dAdo• radical upon reductive cleavage of SAM (state 1 → 2). The C5′ radical position (magenta) is situated within the X-ray crystal structure (state 0) based on ENDOR constraints. The rest of the 5′-dAdo• radical is simply shifted from cluster bound SAM (state 0, shown in gray).
RESULTS
EPR Measurements and Analysis for Stochastic 1H ENDOR for anAdo• Radical
To help refine the analysis of the anAdo• radical properties, we collected 35 GHz echo-detected EPR spectra and stochastic CW 1H ENDOR spectra, of the radical presented in Figure S1. The simulation parameters that best fit the 1H ENDOR spectra (Table S1) differ only minimally from those previously reported.16
Substrate 13C ENDOR Measurements
The 35 GHz Pulsed Davies 13C ENDOR spectra of LAM with anSAM and PLP linked to isotopically labeled lysines (Chart 1;[u-13C]-L-lysine, [1,2-13C]- and [2-13C]- D/L-lysine) are shown in Figure 3.
Chart 1.

Figure 3.

Davies 13C ENDOR for LAM with anSAM, PLP and substrate with simulation spectrum. Spectra were collected at the radical peak. The baseline spectrum from unlabeled Lys has been subtracted for all Davies spectra. Upper: u-13C Lys, with assignments derived from spectra of isotopologues. Simulation, upper, in blue calculated with parameters: Aiso = 4.5 MHz, 2T = 2.0 MHz, ENDOR line-width, 0.35 MHz. Lower: as described in text using partially labeled Lys (Chart 1). Experimental conditions: MF = 34.6–34.8 GHz, MW pulse length (π) = 120 ns, τ = 600 ns T = 2 K.
These spectra were obtained by subtracting the spectrum of a sample prepared with natural-abundance lysine, which exhibits a broad signal at δν ~ −3 MHz attributable to the ν+ partner of the 14N double-quantum transitions (Figure S2 (gray)), and then normalizing the electron spin echo amplitudes.
The spectrum for uniformly labeled Lys consists of two 13C ENDOR doublets, one with couplings (splittings) ranging from ~3 to ~8 MHz, the other with a weak coupling of ~0.4 MHz. The larger coupling is shown to be associated with 2-13C by its persistence in the samples prepared with [1,2-13C]- and [2-13C]-lysine (Figure 3, lower). The weak coupling is associated with 1-13C, as shown by its persistence in the [1,2-13C]-Lys sample and its negligible presence in the [2-13C]- sample (Figure S3, S4); the analysis of the 2-C response is given in the next subsection; that of 1-C is given in SI.44
Interestingly, the intensity of both ENDOR signals in Figure 3, which are normalized to the EPR echo intensity from the anAdo• radical formed by anSAM cleavage, is larger in the spectra with [u-13C]-L-Lys, compared to those with D/L-Lys [1,2-13C] and [2-13C]. We interpret this to mean that anSAM is cleaved in ternary complexes with both D- and L-Lys, but that L-Lys is bound preferentially, and with the 2-13C of D-Lys farther from the anAdo• radical. This inference is derived from the spectra of Figure 3, lower and the expansion of the frequency range, ±2 MHz in Figure S5. These spectra show that the ENDOR responses for both D/L-Lys [1,2-13C] and [2-13C] exhibit an extremely weak, sharp doublet with an intermediate coupling (Aobs = ~ 2 MHz), that we may assign to 2-13C of D-Lys bound in a nonproductive conformation, farther from C5′ of the anAdo• radical.
Analysis of 2-13C ENDOR Response
The shape of the [2-13C]-Lys signal is typical of a powder ENDOR “Pake pattern” (see Materials and Methods) in which an axial anisotropic coupling (T = [−T, −T, +2T]) is combined with an isotropic coupling (Aiso), where T and Aiso have the same sign; as it is expected that T is dominated by through-space dipolar interactions between spin on C5′ and 2-13C with T > 0, this implies that Aiso > 0. The sign of the 2-13C hyperfine coupling constants is confirmed by pulsed ENDOR saturation and recovery (PESTRE) measurements (Figure S6).
In such a Pake pattern spectrum, the splitting between the intense “perpendicular” peaks of the two branches of the spectrum is A⊥ = (Aiso − T), that between the extremal shoulders, is A|| = (Aiso + 2T), and the width of each branch is 3T/2. Simulation of the spectrum gives Aiso = +4.5 MHz and T = +1.0 MHz. As the EPR and ENDOR measurements of anAdo• radical confirm that this radical has not formed a covalent bond to Lys, the observation of a substantial isotropic coupling to the noncovalently bonded 2-13C (Cα) of Lys therefore is remarkable. Such couplings are normally found only when spin is transferred along a covalent pathway between electron spin and coupled nucleus. We show below that this finding instead is the consequence of a tight van der Waals contact between C5′ of anAdo• radical and 2-13C (Cα) of Lys.
The anisotropic coupling matrix in principle is the sum of two axial matrices, the nonlocal contribution (Tn-loc) from through-space dipolar interaction between the electron spin of anAdo• radical and the 13C nuclear spin, and a local contribution (Tloc), caused by interaction of electron spin density delocalized into the C–H bonds of 2-C of Lys: T = Tloc+ Tn-loc. Assuming the latter term comes from the point-dipolar interaction between 2-13C nucleus and the spin density on C5′ (ρ = 0.59; SI), the distance from C5′ to 2-13C of lysine, r, is estimated (see SI) to be r ~ 2.7 Å. However, the point-dipole treatment underestimates the interaction at short distances: the spin on C5′ is not confined to a point, but is distributed within its 2pπ orbital.42 Application of the McConnell–Strathdee correction to the highly resolved axial 2-13C ENDOR pattern (Figure 3) indicates that 2-C lies along the axis of the 2pπ C5′ orbital, as illustrated in Figure 4;45 if 2-C lay appreciably off the π-orbital axis, the anisotropic coupling would acquire a rhombic component absent here. Taking 2-C to lie precisely on-axis for concreteness, the correction yields a C5′ ↔ 2-C distance of ~3 Å,42 satisfyingly in agreement with the van der Waals distance expected when a 2pπ-orbital contact is involved.
Figure 4.

“Cone model” for ENDOR-derived constraints on the distances between C5′ of the anAdo• radical and the carbons and deuterons of Lys. The anAdo• 2pπ orbital is orienated towards the 2-C of Lys. Following the practice of McCracken and co-workers,45 the direction of a C5′-nucleus vector is shown as lying roughly within the cone associated with the nucleus, and similarly for the 3-C–D vector. All distances are shown in Table 1.
Pauli Spin Delocalization
Given the absence of a covalent bond between anAdo• radical and lysine, the presence of a significant isotropic hyperfine coupling to the 2-C (Cα) of substrate lysine, Aiso = +4.5 MHz, might appear surprising, as such couplings normally arise from through-bond spin delocalization. However, such spin transfer across a noncovalent contact is well documented. The paradigm for such spin transfer from a paramagnetic center to a closed-shell neighbor not linked by a covalent bond was provided long ago by H atoms incorporated in a noble gas (e.g., Kr) matrix, which shows a strong hyperfine coupling to matrix nuclei in the absence of covalent bond formation.46,47
The analysis of H-atom to noble gas-atom spin transfer48 considered the consequences of a tight van der Waals contact between a paramagnetic center and a closed-shell neighbor. In such a structure, the electron charge cloud of the paramagnet’s spin-bearing orbital overlaps the electron charge cloud(s) of the doubly occupied orbitals of the closed-shell neighbor, and a simple description of the electronic structure in terms of orbitals localized on only one or the other of the two centers violates the Pauli exclusion principle. Such overlap can, however, be properly described by requiring that the one-electron orbital containing the unpaired electron, in this case the C5′ 2pπ orbital (|2pπC5′〉) of the anAdo• allyl radical, be orthogonalized to the one-electron orbital(s) of the closed-shell neighbor, in this case 2-C.48 This process delocalizes the spin-bearing orbital onto the closed-shell neighbor, 2-C: the spin-bearing C5′ 2pπ orbital (|2pπC5′〉) acquires a small contribution from sp3 orbitals of 2-C (|sp3〉i), and this “Pauli delocalization” introduces an isotropic hyperfine interaction with the closed-shell neighbor. Thus, the presence of an isotropic coupling to 2-13C reveals that this substrate atom is in tight van der Waals contact with the C5′ π atomic orbital of the spin-bearing allyl, π molecular orbital on the anAdo• radical.
This delocalization of the odd electron results in a hyperfine coupling for 2-13C that can be approximated as
| (6) |
where ρ2s is the spin density transferred into the 2s orbital of Lys 2-C, the sum is over the four sp3 orbitals of 2-C, S(2pπ, sp3)i is the C5′(2pπ) ↔ 2-C(sp3)i overlap integral, ρ(C5′) = +0.59 is the spin density in C5′(2pπ), the factor of 4 reflects the 2s contribution to the 2sp3 hybrid orbitals on 2-13C, and N is the normalization factor for the orthogonalized orbital. Insertion of A(2-13C) = 4.3 MHz into this formula shows that the actual spin delocalization onto 2-C is extremely small, ρ2s ~ 10−3. The operation of Pauli delocalization is supported by the PESTRE measurements, which show that A(2-13C) > 0, in agreement with the prediction of eq 6.
Mims ENDOR from 2H-Lys
In the conversion of state 2 to state 3 (Figure 1), a hydrogen atom on 3-C of Lys is abstracted by the C5′ radical to generate β-Lys• radical. Thus, the most important distance in state 2 is that between C5′ and the target proton on 3-C. In the X-ray crystal structure of state 0, the distance from the C5′ in SAM to the nearest H on 3-C of Lys is 2.9 Å. This distance is too great to abstract a hydrogen atom from substrate,49,50 so in the formation of state 2, the 5′-dAdo• radical must move toward this hydrogen atom immediately after SAM cleavage, as is definitively shown by the 13C ENDOR results described above.
To determine the distance from the C5′ of anAdo• radical to the substrate hydrogen atoms in state 2, we collected 2H Mims ENDOR of anAdo• radical in samples prepared with variously deuterated lysines (Chart 2; [3,3,4,4,5,5,6,6-2H]-, [2,6,6-2H]-) and unlabeled Lys (Figure 5, inset). The [2,6,6-2H]-Lys, which is not deuterated at the target 3-C site, showed a 2H response with observed breadth of, A(2H) ~ 0.6–7 MHz; when scaled by the ratio of nuclear g-factors, this corresponds to a 1H response, A(1H) ~ 4 MHz. The 13C ENDOR discussed above clearly shows that upon cleavage of anSAM the anAdo• radical moves toward 2-C/3-C, which puts C5′ far from the 6-H, so we may assign the breadth of the [2,6,6-2H]-Lys signal as being determined by the dipolar 2H coupling between spin on C5′ of anAdo• radical to 2-H of Lys, giving a rough distance of about 3.8 Å between C5′ and 2-2H.
Chart 2.

Figure 5.

2H Mims ENDOR of LAM with anSAM/PLP/2H-Lys. Experimental spectrum with [3,3,4,4,5,5,6,6-2H]-Lys (red) overlaid with sum (blue) of the simulations of the 3-2H (black dashed) and 4-2H (gray dashed) signals. Inset: Overlay of experimental spectra for [3,3,4,4,5,5,6,6-2H]-Lys (red) and [2,6,6-2H]-Lys (green) and unlabeled Lys (grey). Simulation in main figure: Point-dipole interaction with 3-2H, 2T = 0.96 MHz, P = [42 42, 84] kHz, ENDOR line width = 0.05 MHz; additional intensity in the frequency range, ±0.4 MHz is assigned to 4-2H, with point-dipole interaction having with 2T = 0.45 MHz and ENDOR line width = 0.05 MHz. Conditions: microwave frequency = 34.77 GHz for [2,6,6-2H]-Lys, 34.61 GHz for [3,3,4,4,5,5,6,6-2H]-Lys, MW pulse length (π/2) = 50 ns, τ = 1000 ns for [2,6,6-2H]-Lys, 500 ns for [3,3,4,4,5,5,6,6-2H]-Lys, and T = 2 K.
The 2H Mims ENDOR spectrum for [3,3,4,4,5,5,6,6-2H]-Lys is much broader, Figure 5, and exhibits two well-defined pairs of shoulders on the “outsides” of the pattern. These shoulders are the quadrupole-split features associated with the maximum dipolar splitting, 2T = 0.96 MHz, by a single target 3-2H nucleus of Lys. A C5′ ↔ 3-2H distance in the reactive state 3 of 2.4 Å is obtained by interpretation of 2T within the point-dipole model, eq 5; incorporation of a McConnell and Strathdee42 correction lowers the distance slightly, to 2.2–2.3 Å. As shown in the figure, the ENDOR response is well reproduced by a simulation that adopts such a dipolar coupling; the quadrupole coupling of a C–2H bond is well-known to have axial symmetry with the unique axis along the bond,51 and the magnitude of the observed quadrupolar splitting used in the simulation implies a large angle between the unique axis of the hyperfine tensor, associated with the C5′-3H vector, and the 3C–3H bond. (see Figure 4). Regardless of the precise details of the analysis, the size of the hyperfine coupling to 3-2H, as manifest simply in the breadth of the spectrum, clearly indicates that the reactive C5′ site of the adenosyl radical achieves direct contact with its target proton upon cleavage of SAM.
Finally, we can comment on the other 2H of the deuterated Lys. Considerations of a model for the full structure of state 5 presented below suggest that ENDOR intensity in the inner part of the ENDOR signal likely arises mostly from a 4-2H, and this is included in the simulation. Given the tetrahedral geometry at 3-C and the close contact between one 3-2H and C5′, then the other 3-2H must be pointed “away” such that its distance to C5′ is so great as to yield an unresolvably small hyperfine coupling.
Distance from C5′ Methyl-13C Methionine of anSAM
A 35 GHz Pulsed Mims 13C ENDOR spectra of LAM with [methyl-13C]-anSAM, Lys and PLP is shown in Figure 6. The spectrum comprises a hyperfine-split doublet of asymmetric peaks in which individual features are unresolved because the line width is comparable to the dipolar coupling and because of Mims holes effects. Because of the poor resolution, simulations yield only a range of possible values for the hyperfine couplings: 0.39 < |Aiso| < 0.70 (MHz); 0.12 < T < 0.43 (MHz) (Figure S7). As with 2-13C, the presence of an isotropic coupling implies van der Waals contact between the spin-bearing C5′ of the anAdo• radical and [methyl-13C]-anSAM. The point-dipolar coupling corresponds to a range in distance between C5′ and methyl-carbon in methionine, 3.0 < r < 4.7 (Å), substantially greater than the distance to C5′, with the favored simulation, shown in Figure 6, yielding a distance of C5′ of the anAdo• radical to the methyl carbon of cluster-bound methionine of r ≈ 3.6 Å. The crystal structure shows that before reductive cleavage of SAM this distance is 2.94 Å. The lengthening of this distance after cleavage is in agreement with the idea that the distance between C5′ of anAdo• radical and the methyl-13C of methionine has slightly lengthened as the radical moved toward the substrate after cleavage of anSAM. The simulation yields a negative value for Aiso, which implies the longer distance involves radical contact with a methyl-H, with spin-polarization of the C–H bond yielding a negative spin density of the C.
Figure 6.

13C Mims ENDOR for LAM with [13C-methyl]-anSAM, PLP and substrate with simulation spectrum. Spectrum collected at radical peak. Experimental conditions: MF = 34.74 GHz, MW pulse length (π/2) = 50 ns, τ = 400 ns for Mims, and T = 2 K. Simulations (blue) with ENDOR line-widths of 0 and 0.28 MHz; parameters, Aiso = −0.61 MHz, T = 0.21 MHz (distance r = 3.6 Å).
Constraints on the Distances from C5′ to other Lys Nuclei
ENDOR measurements have also been carried out on Lys 1-13C and 2-15N, as presented in SI. These spectra, like those for [methyl-13C]-anSAM, yield constraints on the distance from C5′- to these nuclei; they are included in Table 1 and visualized in Figure 4.
Table 1.
| nucleus (N) | C5′ ↔ N distances (Å) (ENDOR) |
|---|---|
| 1-C | >3.2 |
| 2-C | 3.0 |
| 3-C | a |
| 3-H | 2.2–2.3 |
| 2-N | >3.2 |
| methyl-carbon of Met | ~3.6 |
| 2-H | ~3.8 |
Signal not resolved.
DISCUSSION
Radical SAM enzymes use a site-differentiated [4Fe-4S] cluster and SAM to initiate remarkably diverse radical reactions, nearly all of which are initiated by the 5′-dAdo• radical intermediate generated upon the reductive cleavage of SAM.1,2 The 5′-dAdo• radical intermediate is a highly reactive primary carbon radical that has never been directly observed, although numerous lines of indirect evidence support its central role in both radical SAM and B12 reactions.12,13,52 Key mechanistic questions for radical SAM enzymes revolve around how the reactivity of this initial radical is controlled and directed toward a single, well-defined hydrogen atom on substrate, in a reaction that is often both regio-and stereospecific. Because the 5′-dAdo• radical is so reactive, however, it has not been possible as of yet to directly observe it in an enzyme active site.
In the present work, we utilize the functional SAM analogue S-3′,4′-anhydroadenosyl-L-methionine (anSAM, Scheme 3), which contains a 3′-4′ double bond that provides allylic stabilization to the anAdo• radical, in order to directly probe the initial radical intermediate in lysine 2,3-aminomutase. Use of this stabilized anAdo• radical intermediate in conjunction with isotopically labeled Lys substrates and isotopic labeling of the methionine fragment of anSAM shows that the C5′ of SAM/anSAM moves a small distance toward the substrate site of H atom abstraction and away from the cluster-bound methionyl fragment upon anSAM S–C5′ bond cleavage.
Structural Consequences of SAM Cleavage
The ENDOR-derived metrical information for the relative position and orientation of the C5′ carbon of anAdo• radical analogue for the reactive 5′-dAdo• radical bound at the active site of C. subterminale SB4 LAM is summarized in Table 1 and Figure 4. When considered in the context of the crystal structure of C. subterminale SB4 LAM with bound SAM and Lys in precursor state 0, these findings reveal the minimal extent of the active site rearrangements accompanying reductive S–C5′ bond cleavage and H atom abstraction from Lys, and the driving force for that rearrangement.
Of fundamental importance to efficient catalysis is the remarkably intimate positioning of the target H of substrate adjacent to C5′ of SAM in the ternary complex of state 0, a separation of only 2.8 Å (Figure 2(A)). In conjunction with the dense packing of the LAM active site (Figure S10), this highly constrains the movements of 5′-dAdo• radical, and limits possible side reactions on its remarkably short journey to its destined reaction with a 3-H of Lys. To illustrate the efficiency introduced by the compact LAM active site, consider the implications of the two most robust C5′ ↔ substrate distances in state 2 as measured here.
The C5′ ↔ 2-C (Lys) distance in the LAM/SAM/Lys state 0 structure is 4.2 Å, while the S–C5′ bond length in SAM is 1.98 Å. Breaking that S–C5′ bond must force the two atoms apart, to a distance that approximates the sum of their van der Waals radii, ~3.2 Å (assuming 1.8 Å for S and 1.5 Å for the sp2 carbon of C5′).53 Thus, the reductive cleavage of the S–C5′ bond would immediately lengthen the S–C5′ distance by ~1.3 Å, quite likely “ballistically” driving the radical toward substrate. If bond-breaking were accompanied by the “most efficient” concomitant movement of C5′, namely displacement directly toward 2-C relative to a motionless S, no additional movement would be required to achieve the C5′ ↔ 2-C van der Waals contact distance of ~3 Å as found by ENDOR spectroscopy. Correspondingly, the C5′ distance to the actual target, 3-H (Lys), in the LAM/SAM/Lys state 0 structure is 2.79 Å, and C5′ would need to move toward 3-H by only ~0.6 Å, or just half the obligatory elongation of the S–C5′ distance upon reductive cleavage, to achieve the ENDOR-estimated distance in the radical state of ~2.2 Å, indicative of van der Waals contact.
The distance constraints of Figure 4 (Table 1) can in fact be self-consistently combined in a least-squares optimization of the position for C5′ of the anAdo•/5′-dAdo• radical within the LAM/SAM/Lys structure (state 0), as shown in Figure 7. This figure further presents an heuristic representation of state 5 by incorporating a representation of the 5′-dAdo• radical generated by translating the corresponding fragment of SAM so as to place C5′ at the location relative to the substrate Lys as obtained with the ENDOR constraints. Of particular note, in this structure the 2pπ orbital on a trigonalized C3′ of the anSAM allyl radical would indeed point toward 2-C of Lys, as inferred above from the analysis of the hyperfine interaction.
There of course will be other, but we suggest secondary, active site rearrangements that contribute to this placement of the 5′-dAdo• radical relative to the substrate target in LAM. The flexibility of the lysyl tail, in particular, is shown by the fact that the X-ray structure does not have resolved density for atoms 3–6 within the tail (Figure S11). This flexibility, among other factors, must lie at the ability of the active site to accommodate both D-and L-lysine, as noted above, although with a preference for the L-isomer. For completeness, we note that one must expect some conformational changes in 5′-dAdo• radical itself.54 Such refinements to the overall structure of state 5 presented in Figure 7 should be revealed by subsequent molecular dynamics simulations.
Regardless of any possible imperfections in the details, Figure 7 dramatically reveals how the active site of LAM “tames” the highly reactive 5′-dAdo• radical: this “free radical” is never free. As seen in the state 0 structure of LAM (Figure 7) the low steric demands of the radical-generating [4Fe-4S]/SAM construct allows the 3-H target site of L-Lys substrate to bind adjacent to the S–C5′ bond. ENDOR shows that the reductive cleavage of the S–C5′ bond propels the C5′ radical site through a total motion of only ~1.5 Å, essentially equal to the total motion (~1.3 Å) that would be imparted just by S–C5′ bond cleavage and separation of the atoms to the sum of their van der Waals radii.53 This motion moves C5′ into van der Waals contact with the 3-H target of Lys, having crossed a mere ~0.6 Å gap from its target as implied by the state 0 structure (Figures 2, 4). At this stage the radical site is nestled in a highly constrained pocket where its position is enforced by van der Waals contacts with Lys 2-C and 1-C as well. In addition, C5′ remains in contact with the CH3–S of methionine.
Placed in context with our earlier study of states 3 and 4,14 these observations fill in a picture of how the active site exerts “van der Waals control” of small motions throughout the LAM catalytic cycle. The study of state 3 showed that H atom abstraction creates the α-NH2 Lys substrate radical in van der Waals contact with the 5′-dAdoH. Substrate rearrangement forms the β-NH2 product radical in van der Waals contact with the 5′-methyl, state 4, thereby enforcing the reciprocal abstraction of an H atom from the 5′-dAdoH, 5′-methyl by the product radical, to generate β-NH2 Lys product and regenerate the 5′-dAdo• radical, state 5. As noted just above, the constrained environment places the 5′-dAdo• radical in contact with the S of methionine, and thus poised for recombination to regenerate SAM (conversion of state 5 to state 6) to complete the catalytic cycle.
It is important to note that the short C5′ ↔ 2-C (Lys) target distance in the LAM/SAM/Lys state 0 structure is not unique to this enzyme/substrate pair, but rather comparable distances from the SAM C5′ to the target C of substrate are found in the other structurally characterized radical SAM enzymes (Table 2).8,10,55–62 Given that the target H of substrate is bound adjacent to the S–C5′ bond of SAM in this wide array of radical SAM enzymes, it seems likely to us that this mechanism of “van der Waals control” of small motions for taming the 5′-dAdo• radical, and indeed for such control of the entire catalytic cycle of radical reactions, is common throughout the radical SAM enzyme superfamily.
Table 2.
| radical SAM enzyme (PDB ID) | C5′ ↔ target C distances (Å) (X-ray) |
|---|---|
| anSME (4K39) | 4.1 |
| BioB (1R30) | 3.9 |
| BtrN (4M7T) | 3.7 |
| HemN (1OLT) | 4.0 |
| MoaA (2FB3) | 4.7–5.3 |
| NosL (4R33) | 4.1 |
| PFL-AE (3CB8) | 4.1 |
| SPL (4FHD) | 3.9 |
Lessons from LAM about Coenzyme B12 Enzymes
B12-dependent radical enzymes share a common mechanism in which a hydrogen atom and a substituent exchange places on neighboring sp3-hybridized carbon atoms,63 precisely as occurs in the isomerization of Lys by LAM. In both B12 enzymes and radical-SAM enzymes such as LAM, a 5′-dAdo• radical formed by homolytic bond cleavage, S–C5′ in radical-SAM and Co–C5′ in the B12 enzymes, carries out the catalytic conversion of substrate to product. In both cases the catalytic cycle is completed by the reformation of the parent cofactor.
The mechanism we describe here for “taming” 5′-dAdo• radical by proximity to substrate coupled with van der Waals control of its motion is, however, in sharp contrast to the behavior of the coenzyme B12-dependent enzymes. The C5′ bound to Co in the B12 coenzyme necessarily faces into the sterically demanding corrinoid ring, away from any possible substrate, and as a result, necessarily is further screened from the substrate target by the 5′-dAdo moiety, which lies between C5′ and the target, as shown in Figure 2(B). This geometry forces the target site of substrate to lie “far away” from the Co–C5′ bond, restricting access to the target by the C5′ radical formed through Co–C5′ bond cleavage.64 As a result, the radical must migrate long distances to the site of reaction, as explicitly shown in the elegant advanced paramagnetic resonance studies of ethanol-amine ammonia-lyase (EAL) by Warncke,17 LoBrutto,22 and co-workers, as well as studies of the B12-dependent lysine 5,6-aminomutase by Frey, Ke, and co-workers,25 which revealed that C5′ of 5′-dAdo• radical migrates over 6 Å from its location in the Co–C5′ bond to the reactive position adjacent to substrate. The migration of C5′ required for reaction with its substrate in EAL was modeled by keeping the adenine ring of 5′-dAdo• radical essentially fixed, and introducing a ribosyl rotation around the N-glycosidic bond.65 For glutamate and methylmalonlyl-coenzyme A mutases, the C5′ appears to undergo migration of at most only a few Å, and in fact the cob(II)alamin intermediate has been proposed to function as a conductor rather than a mere spectator in these radical rearrangements.66 The smaller movement in these B12 enzymes is achieved by rotation of the entire adenosyl group,67 or by pseudorotation of the ribose moiety from the C2′-endo to the C3′-endo conformation.23,68
Why B12 enzymes?
Iron–sulfur proteins are widely accepted to be evolutionarily ancient, perhaps emerging from prebiotic iron–sulfur minerals that catalyzed the synthesis of key molecular precursors of life.69 The ubiquitous iron–sulfur protein ferredoxin has been described as a living relic of these primordial FeS catalysts.70 At the present moment in evolution, radical SAM enzymes are found throughout the life on earth, with over 100 000 predicted radical SAM enzymes71 and substrates that range from large proteins—such as pyruvate formate lyase, through small molecules—such as lysine, and including key roles in nucleotide chemistry and cofactor biosynthesis.10,52 Cobalamin, a highly complex radical-generating cofactor, in contrast, is thought to have emerged more recently than radical SAM enzymes,12 yet there are only 12 known cobalamin enzymes, catalyzing a much more limited range of reactions. These observations lead to two obvious questions.
The first question is, why have radical SAM enzymes retained their dominance among extant radical enzymes? Certainly, one factor must be that “they got here first”. However, the results reported here suggest that a significant contributing factor is that these ancient enzymes provide a more flexible architecture for carrying out highly controlled radical catalysis on diverse substrates. For most B12 radical enzymes, catalysis requires that the active site control long-distance (~6 Å) migrations of C5′ of 5′-dAdo• radical because steric congestion shrouds the site of radical generation (Figure 2(B)), forcing remote binding of substrate. This may be contrasted with the minimal, roughly 0.6 Å, motion of the C5′ toward the 3-H target of Lys estimated in this study of LAM (Figure 7), which is enabled by the nearly unfettered access of the Lys 3-H substrate target to the radical-generating [4Fe-4S]-SAM machinery of LAM (Figure 2(A)). Available structural data on other radical SAM enzymes points to similar uninhibited access as reported here for LAM (Table 2). We suggest that the low steric constraints of the [4Fe-4S]/SAM construct and resultant accessibility to substrate of the C5′ radical provided a major reason why evolution spread and retained the use of this construct across all of the kingdoms of life, with thousands of family members catalyzing transformations of the widest range of substrates. The contrasting high steric demands of the coenzyme B12 force the target in B12-dependent enzymes to bind further from the site of the nascent C5′ radical, and require the radical to migrate longer distances to the target site. It seems plausible that this has contributed strongly to the restricted use of B12 radical enzymes.
This analysis, however, raises the evolutionary question: with radical SAM enzymes readily available and apparently more amenable to diverse substrates, why did B12 radical enzymes emerge and persist? One possible answer may be that iron–sulfur clusters are notoriously oxygen-sensitive. Perhaps, with the increase in oxygen in the atmosphere after the evolution of oxygenic photosynthesis, cobalamin appeared where oxygen sensitivity and lability of iron–sulfur clusters posed too much of a detriment to the function of radical SAM enzymes. The cobalamin cofactor has the added advantage of being “self-contained”, without the need of an external reductant as required by radical SAM enzymes. Another very intriguing hypothesis has been proposed by Bückel and co-workers, who suggest that the cob(II)alamin intermediate plays an integral role in catalysis in some B12 radical enzymes, namely glutamate and methylmalonyl-CoA mutases, by interacting with and stabilizing both the 5′-dAdo• radical and product methylene radicals.66 Because of this intimate interaction between the cob(II)alamin and radical intermediates, it is argued, other radical-generating systems such as [4Fe-4S]-SAM cannot provide a direct replacement of this reactivity. We anticipate further conversations regarding why and how nature evolved the B12 enzymes, considering that the low steric constraints of radical SAM enzymes lead to their inherent substrate diversity, a property based on the use of [4Fe-4S]-SAM as “the rich man’s adenosyl cobalamin”.52
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Professor Perry Frey for his seminal contributions to the understanding of LAM and of the parallels between B12 and radical SAM enzymes, and for his support as we carried forward this work upon his retirement. The authors acknowledge the contribution of Dr. Susan Veneziano in creating the His-tagged LAM construct. This work has been supported by the NIH (GM54608, JBB; GM 111097, BMH).
Footnotes
The authors declare no competing fiinancial interest.
Additional EPR and ENDOR data and details of analysis are provided. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b00498.
References
- 1.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114:4229. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frey PA, Hegeman AD, Ruzicka FJ. Crit Rev Biochem Mol Biol. 2008;43:63. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 3.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. J Am Chem Soc. 2002;124:11270. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 4.Walsby CJ, Hong W, Broderick WE, Cheek J, Ortillo D, Broderick JB, Hoffman BM. J Am Chem Soc. 2002;124:3143. doi: 10.1021/ja012034s. [DOI] [PubMed] [Google Scholar]
- 5.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. J Am Chem Soc. 2002;124:11270. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 6.Walsby CJ, Ortillo D, Yang J, Nnyepi M, Broderick WE, Hoffman BM, Broderick JB. Inorg Chem. 2005;44:727. doi: 10.1021/ic0484811. [DOI] [PubMed] [Google Scholar]
- 7.Chen D, Walsby C, Hoffman BM, Frey PA. J Am Chem Soc. 2003;125:11788. doi: 10.1021/ja036120z. [DOI] [PubMed] [Google Scholar]
- 8.Vey JL, Yang J, Li M, Broderick WE, Broderick JB, Drennan CL. Proc Natl Acad Sci U S A. 2008;105:16137. doi: 10.1073/pnas.0806640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lepore BW, Ruzicka FJ, Frey PA, Ringe D. Proc Natl Acad Sci U S A. 2005;102:13819. doi: 10.1073/pnas.0505726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vey JL, Drennan CL. Chem Rev. 2011;111:2487. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frey PA. Comprehensive Natural Products II Chemistry and Biology. Vol. 7. Elsevier Science; Amsterdam: 2010. p. 501. [Google Scholar]
- 12.Frey PA. FASEB J. 1993;7:662. doi: 10.1096/fasebj.7.8.8500691. [DOI] [PubMed] [Google Scholar]
- 13.Frey PA. Acc Chem Res. 2014;47:540. doi: 10.1021/ar400194k. [DOI] [PubMed] [Google Scholar]
- 14.Lees NS, Chen D, Walsby C, Behshad E, Frey PA, Hoffman BM. J Am Chem Soc. 2006;128:10145. doi: 10.1021/ja061282r. [DOI] [PubMed] [Google Scholar]
- 15.Magnusson OT, Reed GH, Frey PA. J Am Chem Soc. 1999;121:9764. [Google Scholar]
- 16.Magnusson OT, Reed GH, Frey PA. Biochemistry. 2001;40:7773. doi: 10.1021/bi0104569. [DOI] [PubMed] [Google Scholar]
- 17.Canfield JM, Warncke K. J Phys Chem B. 2002;106:8831. [Google Scholar]
- 18.Canfield JM, Warncke K. J Phys Chem B. 2005;109:3053. doi: 10.1021/jp046167m. [DOI] [PubMed] [Google Scholar]
- 19.Warncke K, Utada AS. J Am Chem Soc. 2001;123:8564. doi: 10.1021/ja003658l. [DOI] [PubMed] [Google Scholar]
- 20.Warncke K, Canfield JM. J Am Chem Soc. 2004;126:5930. doi: 10.1021/ja031569d. [DOI] [PubMed] [Google Scholar]
- 21.Warncke K. Biochemistry. 2005;44:3184. doi: 10.1021/bi048196t. [DOI] [PubMed] [Google Scholar]
- 22.LoBrutto R, Bandarian V, Magnusson OT, Chen X, Schramm VL, Reed GH. Biochemistry. 2001;40:9. doi: 10.1021/bi001865s. [DOI] [PubMed] [Google Scholar]
- 23.Gruber K, Reitzer R, Kratky C. Angew Chem, Int Ed. 2001;40:3377. doi: 10.1002/1521-3773(20010917)40:18<3377::aid-anie3377>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 24.Zhu C, Warncke K. J Am Chem Soc. 2010;132:9610. doi: 10.1021/ja907769g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maity AN, Hsieh CP, Huang MH, Chen YH, Tang KH, Behshad E, Frey PA, Ke SC. J Phys Chem B. 2009;113:12161. doi: 10.1021/jp905357a. [DOI] [PubMed] [Google Scholar]
- 26.Marsh ENG, Meléndez GDR. Biochim Biophys Acta, Proteins Proteomics. 2012;1824:1154. doi: 10.1016/j.bbapap.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roth JR, Lawrence J, Bobik T. Annu Rev Microbiol. 1996;50:137. doi: 10.1146/annurev.micro.50.1.137. [DOI] [PubMed] [Google Scholar]
- 28.Banerjee R. Chem Rev. 2003;103:2083. doi: 10.1021/cr0204395. [DOI] [PubMed] [Google Scholar]
- 29.Toraya T. Chem Rev. 2003;103:2095. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- 30.Brown KL. Chem Rev. 2005;105:2075. doi: 10.1021/cr030720z. [DOI] [PubMed] [Google Scholar]
- 31.Bradford MM. Anal Biochem. 1976;72:248. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 32.Fish WW. Methods Enzymol. 1988;158:357. doi: 10.1016/0076-6879(88)58067-9. [DOI] [PubMed] [Google Scholar]
- 33.Hinckley GT, Frey PA. Biochemistry. 2006;45:3219. doi: 10.1021/bi0519497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathews II, Erion MD, Ealick SE. Biochemistry. 1998;37:15607. doi: 10.1021/bi9815445. [DOI] [PubMed] [Google Scholar]
- 35.Park J, Tai J, Roessner CA, Scott AI. Bioorg Med Chem. 1996;4:2179. doi: 10.1016/s0968-0896(96)00228-3. [DOI] [PubMed] [Google Scholar]
- 36.Werst MM, Davoust CE, Hoffman BM. J Am Chem Soc. 1991;113:1533. [Google Scholar]
- 37.Davoust CE, Doan PE, Hoffman BM. J Magn Reson. 1996;119:38. [Google Scholar]
- 38.Brueggemann W, Niklas JR. J Magn Reson, Ser A. 1994;108:25. [Google Scholar]
- 39.Doan PE, Hoffman BM. Chem Phys Lett. 1997;269:208. [Google Scholar]
- 40.Doan PE. J Magn Reson. 2011;208:76. doi: 10.1016/j.jmr.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doan PE, Lees NS, Shanmugam M, Hoffman BM. Appl Magn Reson. 2010;37:763. doi: 10.1007/s00723-009-0083-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McConnell HM, Strathdee J. Mol Phys. 1959;2:129. [Google Scholar]
- 43.Willems JP, Lee HI, Burdi D, Doan PE, Stubbe J, Hoffman BM. J Am Chem Soc. 1997;119:9816. [Google Scholar]
- 44.A very weak signal in the 2–13C sample is assigned to a small amount of scrambled label.
- 45.Muthukumaran RB, Grzyska PK, Hausinger RP, McCracken J. Biochemistry. 2007;46:5951. doi: 10.1021/bi700562t. [DOI] [PubMed] [Google Scholar]
- 46.Foner SN, Cochran EL, Bowers VA, Jen CK. J Chem Phys. 1960;32:963. [Google Scholar]
- 47.Morton JR, Preston KF, Strach SJ, Adrian FJ, Jette AN. J Chem Phys. 1979;70:2889. [Google Scholar]
- 48.Adrian FJ. J Chem Phys. 1960;32:972. [Google Scholar]
- 49.Hammes-Schiffer S. Acc Chem Res. 2001;34:273. doi: 10.1021/ar9901117. [DOI] [PubMed] [Google Scholar]
- 50.Hatcher E, Soudackov AV, Hammes-Schiffer S. J Am Chem Soc. 2004;126:5763. doi: 10.1021/ja039606o. [DOI] [PubMed] [Google Scholar]
- 51.Lucken EAC. Nuclear Quadrupole Coupling Constants. Academic Press; New York: 1969. [Google Scholar]
- 52.Frey PA, Magnusson OT. Chem Rev. 2003;103:2129. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 53.Bondi A. J Phys Chem. 1964;68:441. [Google Scholar]
- 54.In the analogue to State 5 studied here, with anAdo• instead of Ado•, there must of course be an additional change in the conformation of the ribose ring to accommodate the change from tetrahedral to trigonal geometries at C4′ and C3′, associated with the presence of the allyl radical comprising C3′, C4′, C5′.
- 55.Goldman PJ, Grove TL, Sites LA, McLaughlin MI, Booker SJ, Drennan CL. Proc Natl Acad Sci U S A. 2013;110:8519. doi: 10.1073/pnas.1302417110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Science. 2004;303:76. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldman PJ, Grove TL, Booker SJ, Drennan CL. Proc Natl Acad Sci U S A. 2013;110:15949. doi: 10.1073/pnas.1312228110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Layer G, Moser J, Heinz DW, Jahn D, Schubert WD. EMBO J. 2003;22:6214. doi: 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hänzelmann P, Schindelin H. Proc Natl Acad Sci U S A. 2004;101:12870. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hänzelmann P, Schindelin H. Proc Natl Acad Sci U S A. 2006;103:6829. doi: 10.1073/pnas.0510711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nicolet Y, Zeppieri L, Amara P, Fontecilla-Camps JC. Angew Chem. 2014;126:12034. doi: 10.1002/anie.201407320. [DOI] [PubMed] [Google Scholar]
- 62.Benjdia A, Heil K, Barends TRM, Carell T, Schlichting I. Nucleic Acids Res. 2012;40:9308. doi: 10.1093/nar/gks603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Buckel W, Friedrich P, Golding BT. Angew Chem. 2012;51:9974. doi: 10.1002/anie.201205299. [DOI] [PubMed] [Google Scholar]
- 64.Shibata N, Tamagaki H, Hieda N, Akita K, Komori H, Shomura Y, Terawaki S-i, Mori K, Yasuoka N, Higuchi H, Toraya T. J Biol Chem. 2010;285:26484. doi: 10.1074/jbc.M110.125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khoroshun DV, Warncke K, Ke SC, Musaev DG, Morokuma K. J Am Chem Soc. 2003;125:570. doi: 10.1021/ja028393k. [DOI] [PubMed] [Google Scholar]
- 66.Buckel W, Kratky C, Golding BT. Chem—Eur J. 2006;12:352. doi: 10.1002/chem.200501074. [DOI] [PubMed] [Google Scholar]
- 67.Kim SJ, Lester C, Begley TP. J Org Chem. 1995;60:6256. [Google Scholar]
- 68.Buckel W, Friedrich P, Golding BT. Angew Chem, Int Ed. 2012;51:9974. doi: 10.1002/anie.201205299. [DOI] [PubMed] [Google Scholar]
- 69.Huber C, Wächtershäuser G. Science. 1998;281:670. doi: 10.1126/science.281.5377.670. [DOI] [PubMed] [Google Scholar]
- 70.Eck RV, Dayhoff MO. Science. 1966;152:363. doi: 10.1126/science.152.3720.363. [DOI] [PubMed] [Google Scholar]
- 71.Babbitt P, Gerlt J. [accessed 05/12/15];Structure Function Linkage Database. 2013 http://sfld.rbvi.ucsf.edu/django/superfamily/29/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.