

Pathologic cellular states often result from corruption of existing gene regulatory programs. As such, a major effort is underway to discover regulatory network perturbations underlying disease initiation and progression. The majority of these approaches involve either: (i) comparing the activity of specific candidate regulatory programs, from transcription factors to microRNAs to chromatin states, or (ii) performing a broad agnostic search across the space of all conceivable regulatory interactions to discover the key underlying causal perturbations. Despite its well-documented success, the first approach is nevertheless limited to only the set of regulatory interactions that are known. The second approach, on the other hand, endeavors to discover known or novel regulatory interactions through global analysis of whole-genome measurements. Identification of putative cis-regulatory elements (sequence-motif discovery) is a fitting example. The underlying assumption of this framework is that transcripts behaving similarly across a large set of samples are targeted by the same regulatory factor(s) and share similar cis-regulatory sequence elements in their regulatory regions. Thus, starting from expression datasets, various search algorithms are used to identify the sequence-motifs that are most over-represented in promoters of transcripts displaying coordinated behavior.1 Given the success of this approach in revealing transcriptional regulons, it was subsequently extended to RNA sequences to identify cis-acting elements at the post-transcriptional level. For example, RNA-binding protein and microRNA recognition sites can be successfully identified along the 3′ UTRs of many transcripts. However, it has become increasingly evident that the ability of RNA to form more complex secondary structures drastically affects the functionality of cis-acting elements. Many interacting partners recognize the structural features of the RNA, rather than its linear sequence; in other cases, the local secondary structure determines the accessibility of a specific site for direct interaction. Thus, a comprehensive approach toward identifying post-transcriptional cis-regulatory elements should incorporate structural as well as sequence cues in its search algorithm. Building on prior methods for probing the regulatory sequence and structure of RNA,1,2 we thus developed a new computational framework called TEISER to search the immense space of small RNA structural elements to discover candidate predictions that are significantly informative of global RNA behavior.3
A global unbiased approach such as the one described above can help reveal the cis-regulatory signature of post-transcriptional perturbations underlying disease states. Cancer metastasis is a multistep process wherein cell populations selected for enhanced metastatic capacity coopt a suite of molecular mechanisms to modulate gene expression levels.4 The systematic characterization of metastasis-suppressive and promoting microRNAs has highlighted the potential role of post-transcriptional regulatory programs in cancer metastasis.5,6 As such, we set out to perform an agnostic search for post-transcriptional regulatory programs differentially engaged in highly and poorly metastatic cells to reveal heretofore uncharacterized molecular mechanisms of metastatic disease. We first performed transcriptome-wide mRNA stability measurements using 4-thiouridine pulse-labeling of cellular RNA followed by capture and high-throughput sequencing of labeled RNA at varying time-points.7 We estimated differential transcript stability for roughly 13,000 mRNAs in poorly metastatic breast cancer cells relative to their in vivo-selected highly metastatic derivatives. Application of TEISER to these data revealed a family of GC-rich stem-loops that were significantly enriched in transcripts that are destabilized in highly metastatic cells. Using reporter assays, we showed that these structural elements are sufficient for transcript destabilization. A computational search for RNA-binding proteins whose mRNA expression correlates with the stem-loop regulon identified TARBP2 as a potential upstream regulator. TARBP2, a double-stranded RNA binding protein previously implicated in miRNA processing, is known to interact with stem-loops and is a functional component of the RNAi silencing complex. Silencing TARBP2 followed by gene expression profiling and whole-transcriptome stability measurements indicated that this protein plays a role in modulating the expression and stability of transcripts carrying the stem-loop element. We used cross-linking immunoprecipitation followed by high-throughput sequencing (HITS-CLIP) to assess whether TARBP2 directly interacts with endogenous transcripts in vivo. Our results indicated that: (i) TARBP2 binds GC-rich stem-loops in vivo, (ii) TARBP2-bound transcripts are stabilized in TARBP2-knockdown cells, and (iii) TARBP2-bound transcripts are significantly enriched among those destabilized in highly metastatic cells Fig. 1.
Figure 1.
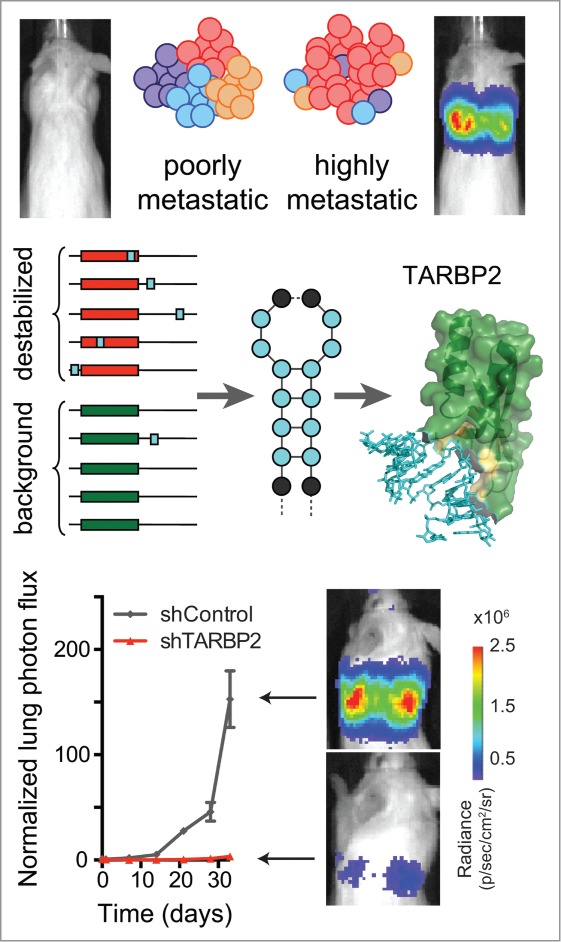
Discovery and functional validation of a TARBP2-mediated post-transcriptional regulatory program. Analysis of differential stability measurements comparing poorly metastatic breast cancer cells to their in vivo-selected highly metastatic derivatives led to the identification of a family of GC-rich stem-loops enriched in transcripts that were destabilized in highly metastatic cells (schematized as light-blue boxes and a generic stem-loop). Subsequent analysis revealed TARBP2 as the post-transcriptional regulator that binds these structural elements in vivo. Consistent with its hypothesized role as a promoter of metastatic progression, TARBP2 depletion by RNAi significantly reduced metastatic burden in mouse lung colonization assays.
Using lung colonization assays in mice, we showed that TARBP2 plays a direct role in promoting metastasis. Silencing of TARBP2 resulted in a substantial reduction in metastatic burden in the lungs of xenografted mice. Follow-up in vitro and in vivo experiments revealed that TARBP2 promotes metastasis by destabilizing ZNF395 and APP (Amyloid precursor protein) transcripts, 2 newly implicated metastasis suppressor genes that were previously associated with Huntington's and Alzheimer's disease, respectively. We also noted a higher expression of TARBP2 in human tumors that metastasized (stage IV) and an association with lower relapse-free survival. In support of our findings, a link between TARBP2 up-regulation and poor prognosis in breast cancer was also recently reported.8 Our results revealed that TARBP2 over-expression enables cancer cells to mount a 2-pronged attack against metastasis suppressive pathways. There are important questions, however, that remain unanswered: (i) how do cancer cells up-regulate TARBP2? (ii) what is the nature of the molecular mechanism through which TARBP2 destabilizes its target transcripts? (iii) are there therapeutic interventions that can disrupt this pathway? These are exciting directions to pursue in future work.
References
- 1. Elemento O, et al. . Molecular cell 2007; 28:337-50; PMID:17964271; http://dx.doi.org/ 10.1016/j.molcel.2007.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Foat BC, et al. . Mol Syst Biol 2009; 5:268; PMID:19401680; http://dx.doi.org/ 10.1038/msb.2009.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goodarzi H, et al. . Nature 2012; 485:264-8.; PMID:22495308; http://dx.doi.org/ 10.1038/nature11013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Minn AJ, et al. . Nature 2005; 436:518-24.; PMID:16049480; http://dx.doi.org/ 10.1038/nature03799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pencheva N, et al. . Cell 2012; 151:1068-82.; PMID:23142051; http://dx.doi.org/ 10.1016/j.cell.2012.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Png KJ, et al. . Nature 2012; 481:190-94; http://dx.doi.org/ 10.1038/nature10661 [DOI] [PubMed] [Google Scholar]
- 7. Goodarzi H, et al. . Nature 2014; 513:256-60; PMID:25043050; http://dx.doi.org/ 10.1038/nature13466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin X, et al. . Med Oncol 2014; 31:868.; PMID:24563327; http://dx.doi.org/ 10.1007/s12032-014-0868-9 [DOI] [PubMed] [Google Scholar]