

Abstract
In this issue of Cancer Cell, Sturm et al. report that global DNA methylation patterns in glioblastoma multiforme divide adult and pediatric tumors into subgroups that have characteristic DNA mutations, mRNA profiles, and most importantly, different clinical behaviors. These findings suggest novel opportunities for therapeutics for this dreaded disease.
Glioblastoma multiforme (GBM) is the most aggressive brain tumor and is associated with very poor overall survival. GBM occurs in adults much more frequently than in children or adolescents, and pediatric GBM has genetic abnormalities that make it distinct from adult tumors, suggesting that although the microscopic appearance and grim prognosis are shared (Figure 1), pediatric and adult GBM have different underlying biologies (Paugh et al., 2010). In this issue of Cancer Cell, Sturm et al. (2012) show that pediatric GBM contains two epigenetic subgroups that are distinct from those found in adult tumors. The epigenetic profiles of these groups correlate tightly with mutations in H3F3A. This gene encodes the replication-independent histone H3.3, which predominantly binds transcriptionally active loci and telomeres. H3.3 is frequently methylated at or near the residues that are mutated. Alterations in histone methylation affect the accessibility of the associated DNA and may promote changes in DNA methylation (Turcan et al., 2012). H3F3A mutations therefore provide a potential mechanism underlying the global methylation changes observed in these pediatric GBM subgroups.
Figure 1. Similar Appearances, Different Methylomes, and Divergent Outcomes.
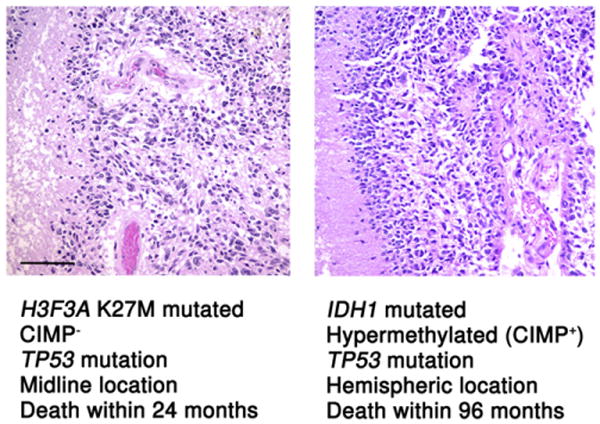
A pediatric midline GBM of the pons (a diffuse intrinsic pontine glioma, left) and a hemispheric glioblastoma multiforme arising in a young adult (right) have a similar histologic appearance on high power hematoxylin and eosin photomicrographs. Yet, as demonstrated by Sturm et al. (2012) in this issue of Cancer Cell, these two tumors affect different patient populations, arise in anatomically distinct regions, have different methylation profiles, and behave differently. For both images, magnification = 200× and scale bar = 100 μM.
These findings build on earlier studies that showed that adult GBM could be subdivided into three epigenetic subgroups, one of which correlated with mutations in the metabolic gene IDH1 (Noushmehr et al., 2010; Parsons et al., 2008; Verhaak et al., 2010). Patients without IDH1 mutation are older and have more rapidly progressive disease, whereas those with IDH1 mutation frequently have an antecedent low grade glioma, are younger, have more frequent TP53 mutations, and are less likely to have receptor tyrosine kinase amplification (Parsons et al., 2008). The IDH1 mutation is a gain of function alteration that creates a novel onco-metabolite, 2-hydroxyglutarate (2HG), which interferes with the normal cellular methylation machinery. This in turn leads to widespread increases in global methylation known as the CpG-island methylator phenotype (CIMP) (Turcan et al., 2012). mRNA expression profiling identified four subgroups of adult GBM (proneural, neural, classical, and mesenchymal) and determined that each subgroup contains distinct pathway alterations (Verhaak et al., 2010). Tumors with IDH1 mutations fall primarily into the proneural expression profile, and in epigenetic analyses, CIMP+ tumors segregate into that same group (Noushmehr et al., 2010).
IDH1 is rarely altered in pediatric GBMs. Indeed, pediatric GBM contain significantly different genomic alterations from adult tumors (Paugh et al., 2010). The recent discovery that mutations in the histone gene H3F3A occur predominantly in pediatric high grade gliomas (Schwartzentruber et al., 2012) further emphasizes the differences between adult and pediatric GBM. Sturm et al. (2012) now show that an expanded analysis of the methylome of GBM, which includes pediatric GBM cases, can divide this tumor into six subgroups, two of which are primarily pediatric, and the remainder of which are primarily adult. Methylation groups largely correlate with the pattern of expression profiling that was previously reported, emphasizing the importance of epigenetic regulation in GBM (Verhaak et al., 2010).
Sturm et al. (2012) show that three of the epigenetic subgroups (two pediatric and one adult) correlate with mutations in H3F3A and IDH1. H3F3A and IDH1 mutations are non-overlapping, suggesting that they represent different paths to achieve widespread alterations in genomic methylation. The two mutations in H3F3A are seven amino acid residues apart and are associated with dramatically different methylation profiles. The H3F3A G34 mutation is associated with a hypomethylation phenotype, which is most prominent at the ends of chromosomes. The H3F3A K27 mutation is associated with a methylation pattern that is distinct from both that of H3F3A G34 and the CIMP+ phenotype associated with IDH1 mutation.
There is also increasing evidence that alterations in H3F3A and IDH1 interfere with the normal differentiation of neural progenitors. The abnormal metabolite 2HG produced by mutant IDH1 leads to increased repressive methylation at H3K27 and inhibits immortalized neural cell differentiation, suggesting a potential common mechanism for tumorigenesis in IDH1 and H3F3A K27M mutant groups (Lu et al., 2012). IDH1 mutation also leads to increased neural stem cell marker expression and decreased expression of mature differentiation genes in response to differentiation signals, which are hallmarks of pre-cancerous cells (Turcan et al., 2012). Sturm et al. (2012) show in H3F3A G34 mutated glioblastoma a similar decrease in expression of OLIG2, an important neuro-developmental gene. Loss of OLIG2 is associated with increased methylation at the OLIG2 locus, which occurs despite the global hypomethylation of the genome in H3F3A G34 mutated glioblastoma. Although there are intriguing similarities in the pathways between IDH1 and H3F3A mutated glioblastoma, the disparate methylation signatures and clinical course of these three groups involving children and young adults suggest that the mechanism by which these alterations lead to transformation of normal cells will require extensive study.
Although this study shows that different GBM methylation subgroups have somewhat variable clinical courses, the overall outcomes of children and adults with GBM is extremely poor, with few long-term survivors reported in the literature (Louis et al., 2007). It remains to be seen how identification of methylation alterations will translate into improved therapeutic options for these patients. The significant changes in global methylation patterns identified by Sturm et al. (2012) make epigenetic altering agents an attractive therapeutic modality. The Children's Oncology Group is currently investigating altering chromatin structure with histone deacetylase inhibitors in GBM (NCT01236560). However demethylating agents have not been widely investigated in pediatric GBM.
Sturm et al. (2012) show that the G34 subgroup of pediatric GBM is hypomethylated preferentially at the chromosome ends and features the alternative lengthening of telomeres (ALT) phenotype. Although the precise mechanism of ALT remains unknown, the presence of promyelocytic leukemia bodies and evidence of heterologous recombination between telomeres provide enticing targets for therapy (Heaphy et al., 2011). The recent increases in our understanding of the genetics of GBM, and in particular, the integration of both pediatric and adult tumors into an overall epigenomic classification scheme by Sturm et al. (2012), set the stage for translation of molecular advances into improved care for patients with this disease.
References
- Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E, Netto GJ, Epstein JI, Lotan TL, Westra WH, et al. Am J Pathol. 2011;179:1608–1615. doi: 10.1016/j.ajpath.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. Lyon, France: IARC Press; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Cancer Genome Atlas Research Network. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, et al. J Clin Oncol. 2010;28:3061–3068. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fonte-basso AM, Quang DA, Tönjes M, et al. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DTW, Konerman C, Pfaff E, Tonjes M, Sill M, Bender S, et al. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. this issue. [DOI] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Cancer Genome Atlas Research Network. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]