

Abstract
The differential diagnosis of acute leukoencephalopathy often focuses on central nervous system idiopathic inflammatory demyelinating diseases (IIDDs) such as multiple sclerosis (MS). However, a spectrum of conditions mimic IIDDs, therefore it is critical to consider whether symptoms, signs, imaging and/or response to therapies are compatible with the diagnosis. We describe a 32-year-old previously healthy woman presenting with a 2 year history of steroid-responsive relapsing episodes lasting 2–10 days characterized by transient visual blurring, right-hemiparesis, and spells of aphasia. MRI demonstrated multifocal, relapsing, predominantly white matter enhancing brain lesions, a longitudinally extensive cord lesion, and abnormal visual evoked potentials. Notably, some lesions persistently enhanced whereas others demonstrated progressive T2W hypointensity. Brain biopsy revealed an atypical plasma cell infiltrate and crystal-storing histiocytosis, which by mass spectrometry confirmed the presence of macrophages containing intracellular kappa-light chain restricted crystals. Bone marrow was negative. The patient did well for several years on pulse dexamethasone, however subsequent scans demonstrated increasing enhancement. Repeat biopsy demonstrated a clonal plasma cell proliferation. She was treated with melphalan, and has remained stable. Although this patient initially met McDonald criteria, atypical imaging prompted further workup, and advanced proteomic technology helped secured an accurate diagnosis. Crystal-storing histiocytosis should be considered in the differential diagnosis of inflammatory CNS disorders.
Keywords: Crystal storing histiocytosis, Plasma cell neoplasm, CNS demyelinating disease, Inflammatory CNS disorder, Acute leukoencephalopathy, T2-weight hypointensity
1. Introduction
Although the major consideration in the differential diagnosis of an acute leukoencephalopathy is a CNS IIDD, a broad spectrum of demyelinating and non-demyelinating disorders can mimic IIDD (Weinshenker and Lucchinetti, 1998) and may even fulfill established diagnostic criteria for MS. CSH is a rare pathology associated with disorders that express monoclonal immunoglobulins, such as multiple myeloma, monoclonal gammopathy of undetermined significance, and lymphoid disorders such as lymphoplasmacytic lymphoma or extramedullary plasmacytoma (Lebeau et al., 2002). It is presumed to be due to an intralysosomal accumulation of secreted paraproteins or immunoglobulins, which aggregate into crystals. Isolated CNS involvement is extremely uncommon, with only two prior cases reported in the literature. Although CSH shows a variety of appearances sometimes obscuring the underlying disorder, immunoglobulins of light-chain type kappa (κ) have been almost exclusively involved without a consistent association with a particular heavy chain. This suggests that CSH results from the storage of crystals produced by plasma cell tumors that either overproduce κ-light chain or express a structurally aberrant molecule. We describe a rare case of CNS restricted CSH presenting as a relapsing acute leukoencephalopathy. Atypical clinical and radiographic presentation led to brain biopsy, and diagnosis was confirmed using advanced proteomic technology.
2. Case report
A 32-year-old woman initially presented in February 2004 with recurrent headaches in the setting of right eye visual blurring and subtle incoordination. By April 2004 the headaches intensified, and she had a two-week transient episode of right sided weakness and aphasia. MRI revealed multiple, predominantly white matter, enhancing and nonenhancing lesions with associated edema and heterogenous T2W signal intensity (Fig. 1(A)–(F)). Visual evoked potentials showed right conduction slowing and optic atrophy on funduscopic exam. CSF was normal. Brain biopsy demonstrated inflammation and “crystal deposition” (Fig. 2(a)). Clinical and radiographic improvement followed a course of high dose steroids. In August 2005 she had a recurrent two-week episode of aphasia associated with episodic right-sided jerking. Follow-up MRI revealed persistent enhancement of the left hemispheric lesion with development of new brain lesions. Repeat brain biopsy showed “crystal deposition” (Fig. 2(b)). She again improved to baseline with steroids and phenytoin. Between November 2005 and May of 2006 she continued to experience recurrent transient episodes of aphasia, confusion, and incoordination lasting days to weeks refractory to 6 months of mycophenolate at a dose of 1000 mg twice a day. MRI of the spine in May 2006 (Fig. 1(G)) revealed several T2W lesions throughout the cord from C2-5; T1-4, and T9–T11, with enhancement at C4-5. Extensive workup for inflammatory and metabolic causes was negative. Bone marrow biopsy/aspirate, fat aspirate and CT chest/abdomen/pelvis were normal. Liquid chromatography–electrospray tandem mass spectrometry (LC-MS/MS) performed on the two prior biopsy samples demonstrated κ-light chain deposition. In situ hybridization labeled monotypic κ-plasma cells. Her course stabilized with lenalidomide (Revlimid) and dexametasone. However, by April 2007 she developed recurrent seizures and follow-up brain MRI demonstrated increase in size and extent of enhancing lesions, particularly involving the left frontal lobe, with more prominent T2 hypointensity and new linear T2W hypo-intensities along the lateral ventricles (Fig. 1(H)–(K)). Bone marrow biopsy and PET scan were negative. Repeat brain biopsy yielded a more extensive sampling of the tissue, and revealed a marked increase in plasma cells with plasmablastic features, positive for kappa light chains. The presence of increased cytologic atypia in these cells, compared to previous biopsies, suggested the presence of malignant transformation (Fig. 3). CSF similarly confirmed an increase in κ-light chains. She was treated with melphalan in conjunction with blood–brain barrier disruption utilizing 25% mannitol administered intra-arterially (two 10 mg/M2 treatments/month for 12 months) followed by low dose brain irradiation (3600 cGy over one month) (Fig. 1(L)–(O)). Melphalan was selected because it is used to treat systemic plasma cell malignancies and can be given safely intra-arterially after osmotic blood–brain barrier disruption. Treatment was completed September 2009. She improved clinically and radiographically with treatment and has remained stable for the past two years off of therapy.
Fig. 1.
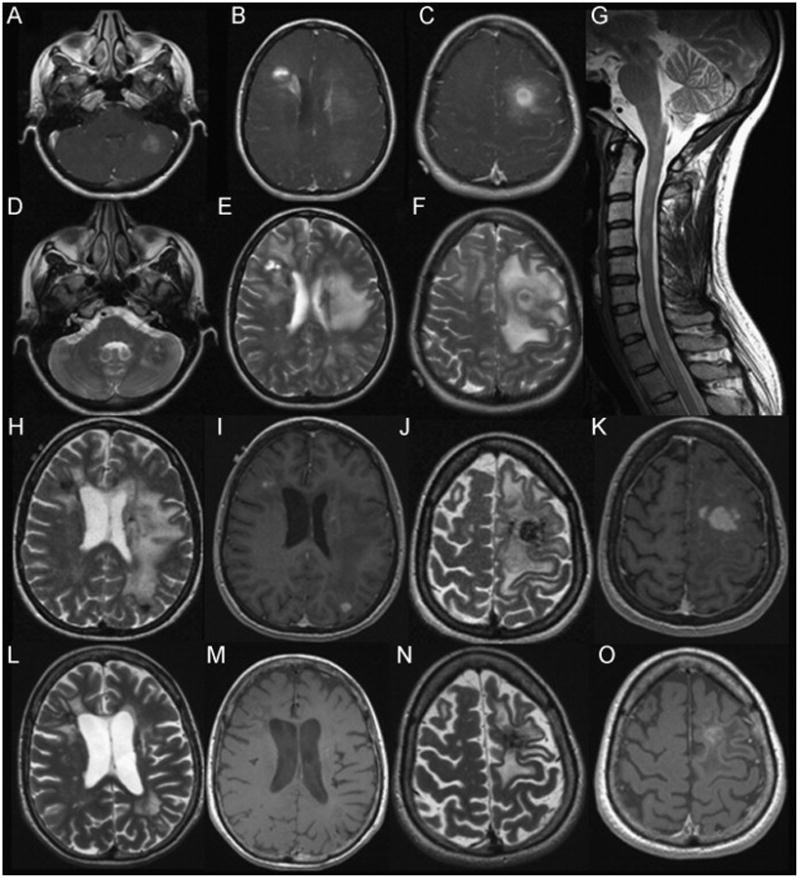
MRI scans from 2004 to 2010. (A)–(F): MRI Brain 4/04. (A)–(C): Ax T1W with Gadolinium demonstrates multiple ill-defined enhancing masses. (D)–(F): Ax T2W sequence demonstrates heterogeneous signal intensity with concentric ring-like pattern of variable T2 hypointensity surrounding a core of heterogeneous signal intensity. Left frontal lesion associated with vasogenic edema, sulcal effacement and slight midline shift (E/F). (G): MRI C-spine 5/06. Sagittal T2W image demonstrates multiple ill-defined cervical and upper thoracic cord lesions. (H)–(K): MRI Brain 4/07. H/J: Ax T2W scans show disease progression with more prominent T2W hypointensity compared to 2006, including areas adjacent to the left lateral ventricle, subcortical right frontal and left parietal lobes and left frontal lobe. I/K: Ax T1W with Gad shows new areas of enhancement as well as persistent enhancement of the left frontal mass. (L)–(O): MRI Brain 5/2010. (L)/(N): Ax T2W scans demonstrate reduced edema, confluent periventricular lesions as well as focal areas of T2W hypointensity. (M)/(O): Ax T1W with Gad demonstrates interval reduction in enhancement of left frontal lesion, with no other enhancing lesions identified.
Fig. 2.
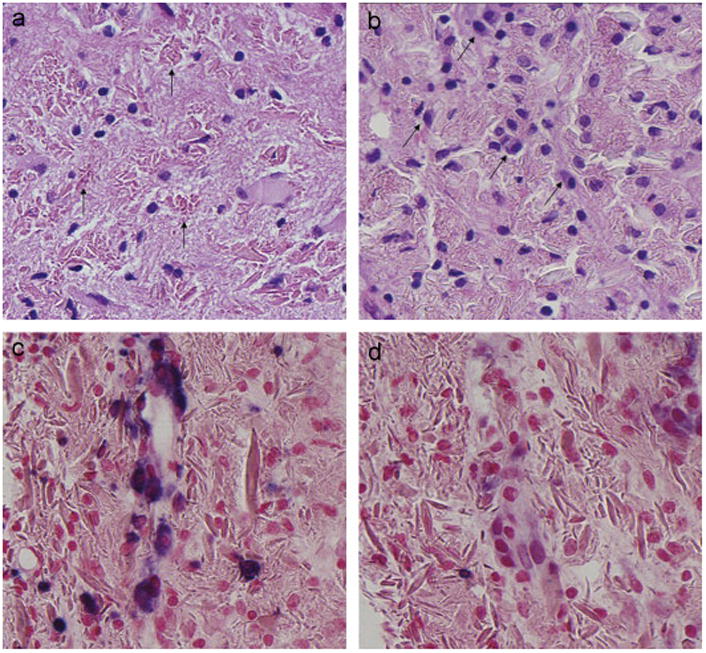
Initial Pathology. (a) Brain biopsy 4/04. Initial brain biopsy demonstrates rod-shaped to rhomboid crystals within macrophages (arrows). (b)–(d) Brain biopsy 2005: A subsequent biopsy showed similar findings with a subtle increase in associated plasma cells (arrows) (b). In situ hybridization using probes targeting light-chain mRNA confirmed κ-light chain restriction (c). Conversely, lambda was negative in most plasma cells (d).
Fig. 3.
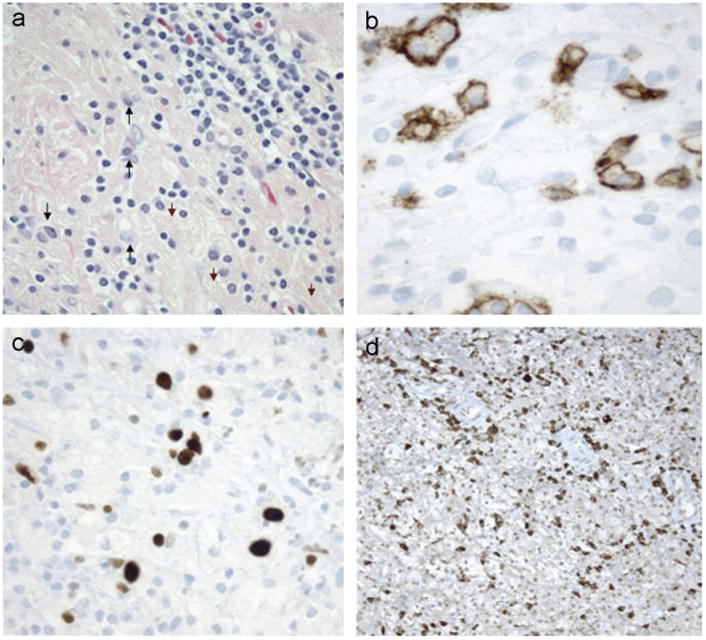
Biopsy Confirming Malignant Transformation. (a)–(d) Brain biopsy 5/07. A third brain biopsy demonstrates an increase in atypical plasma cells (arrows) with crystal laden macrophages (arrowheads) and lymphocytes (a). CD138 is positive in plasma cells with enlarged, atypical nuclei (b). Ki67 demonstrates proliferation of atypical lymphoplasmacytes with enlarged nuclei (c). Immunocytochemistry for κ-light chains highlights many lymphocytes/plasma cells (d). Findings are consistent with plasma cell neoplasm associated with CSH.
3. Discussion
We describe a rare and unique case of CNS restricted CSH associated with a plasma cell malignancy. The initial objective was to evaluate whether the acute leukoencephalopathy represented an IIDD of the CNS versus another inflammatory disorder based on the setting, symptoms, mode of onset, imaging and/or response to therapies. Although the patient had a multifocal, initially steroid responsive, relapsing–remitting predominantly white matter CNS disorder involving brain, spinal cord and optic nerves, the atypical clinical features, rapid progression and atypical presentation, coupled with persistent enhancement and progressive T2W hypointensity on MRI argued against a diagnosis of demyelinating disease, such as MS. Interestingly, the opticospinal involvement, including evidence of a longitudinally extensive myelitis, raised the possibility of a diagnosis of a neuromyelitis optica spectrum disorder. The patient ultimately required brain biopsy to confirm the diagnosis.
Deposition of crystals within histiocytes is an uncommon and unusual pathologic response in the brain, occasionally found in reactive or neoplastic plasma cells, or in the renal tubular epithelium of patients with multiple myeloma and in CNS vasculitis (Lebeau et al., 2002). Only two previous cases of CSH with isolated brain involvement have been reported in the literature. The presence of cerebral polyclonal crystallized Ig deposits and isolated CNS angiitis were described in a 38 year-old women who presented with intractable seizures (Pezeshkpour et al., 1985). She experienced sustained clinical remission following treatment with cyclophosphamide. In a second case, a 27 year-old woman with Crohn's disease presented with asymmetric tremor and left sided sensory disturbance in the setting of a unifocal gadolinium enhancing brain lesion associated with mild mass effect and edema. Biopsy confirmed the presence of intracytoplasmic crystal inclusions within macrophages (Kaminsky et al., 2011). No treatment was mentioned. In both cases a bone marrow to exclude systemic malignancy was not reported. Our patient had similar clinical and radiologic features as the previously reported cases. After the malignant transformation, she responded well to treatment with intra-arterial melphalan following blood–brain barrier disruption and low dose brain irradiation.
The intralysosomal accumulation of crystalline aggregates can present diagnostic difficulties, especially when bone marrow is normal. Other then crystal-storing histiocytosis associated with underlying hematologic malignancies, the monoclonal proteins can aggregate and deposit systemically in light-chain amyloidosis, monoclonal immunoglobulin deposition disease or monoclonal cryoglobulinemia (Lebeau et al., 2002). Few genetic disorder associated with systemic crystal deposition may also affect the CNS including cystinosis or oxalosis. A case report described a man with cystinosis who at age 29 years developed a myelopathy in the setting of contrast-enhancing lesions in the brainstem and cervical cord on MRI. Spinal cord biopsy showed lymphocytic vasculitis and abundant perivascular cystine crystals birefringent under polarized light (Berger et al., 2009). A second case of primary oxalosis reported the presence of calcium oxalate crystals in all tissue examined at post mortem, including brain and meninges. Oxalate crystal deposition has also been observed in cerebral vessels following ethylene glycol ingestion, a condition associated with brain inflammation and edema.
Despite an extensive search for an underlying systemic disorder, this patient's disease remained CNS restricted from onset. Extensive metabolic testing was negative as was systemic workup for hematologic malignancy including a negative bone survey, normal peripheral blood flow cytometric immunophenotyping and normal bone marrow aspirate and biopsy. Furthermore, she had no features to suggest amyloidosis.
Recently, Rodriguez et al. (2008) described a series of 13 cases pathologically characterized by evidence of proteinaceous aggregates deposited in the CNS and utilized a novel method of LC-MS/MS to identify proteins contained within laser microdissected areas of interest. This study emphasized the novel application of mass spectrometry to analyze and identify the content of proteinaceous deposits in the CNS, thereby enhancing the diagnostic precision afforded by routine histology and immunochemistry. Our case was analyzed in this series, and κ-light chains were found in the macrophage intracellular crystals by LC-MS/MS (Rodriguez et al., 2008). CSH typically does show a tendency to κ-light chain restriction, usually due to over-production by plasma cells or sequence abnormalities that promote crystallization or impair lysosomal degradation. Given the association of κ-light chain restriction with lymphoproliferative and plasma cell proliferative disorders, a careful systemic workup and close follow-up were warranted in our patient.
In conclusion, our patient initially met McDonald criteria for diagnosis of MS, but the atypical clinical presentation and neuroimaging prompted further workup, and advanced proteomic technology helped confirm an accurate diagnosis. Although brain CSH is a rare disorder, this case underscores this entity should be considered in the differential diagnosis of acute leukoencephalopathies. Ultimately the diagnosis should be the one that best resolves the clinical, imaging, pathological and laboratory features.
Abbreviations
- CNS
central nervous system
- IIDDs
idiopathic inflammatory demyelinating diseases
- CSH
crystal storing histiocytosis
- MS
multiple sclerosis
- LC-MS/MS
liquid chromatography-electrospray tandem mass spectrometry
Footnotes
Conflict of interest: All the authors have no disclosures related to this manuscript.
References
- Berger JR, Dillon DA, Young BA, Goldstein SJ, Nelson P. Cystinosis of the brain and spinal cord with associated vasculopathy. Journal of the Neurological Sciences. 2009;284(1–2):182–5. doi: 10.1016/j.jns.2009.03.026. [DOI] [PubMed] [Google Scholar]
- Kaminsky IA, Wang AM, Olsen J, Schechter S, Wilson J, Olson R. Central nervous system crystal-storing histiocytosis: neuroimaging, neuropathology, and literature review. American Journal of Neuroradiology. 2011;32:E26–8. doi: 10.3174/ajnr.A1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeau A, Zeindl-Eberhart E, Muller EC, et al. Generalized crystal-storing histiocytosis associated with monoclonal gammopathy: molecular analysis of a disorder with rapid clinical course and review of the literature. Blood. 2002;100(5):1817–27. [PubMed] [Google Scholar]
- Pezeshkpour G, Stuart TD, Estridge MN. Crystalline encephalopathy: cerebral immunoprotein deposits and isolated angiitis. Annals of Neurology. 1985;17:96–9. doi: 10.1002/ana.410170120. [DOI] [PubMed] [Google Scholar]
- Rodriguez FJ, Gamez JD, Vrana JA, et al. Immunoglobulin derived depositions in the nervous system: novel mass spectrometry application for protein characterization in formalin-fixed tissues. Laboratory Investigation. 2008;88:1024–37. doi: 10.1038/labinvest.2008.72. [DOI] [PubMed] [Google Scholar]
- Weinshenker BG, Lucchinetti CF. Acute leukoencephalopathies: differential diagnosis and investigation. The Neurologist. 1998;4:148–66. [Google Scholar]