

Abstract
Background
To review the inflammatory sequelae of traumatic brain injury (TBI) and altitude exposure and discuss the potential impact of aeromedical evacuation (AE) on this process.
Methods
Literature review and expert opinion regarding the inflammatory effects of TBI and AE.
Results
Traumatic brain injury has been called the signature injury of the current military conflict. As a result of the increasing incidence of blast injury, TBI is responsible for significant mortality and enduring morbidity in injured soldiers. Common secondary insults resulting from post-traumatic cerebral inflammation are recognized to adversely impact outcome. AE utilizing Critical Care Air Transport Teams has become a standard of care practice following battlefield injury, to quickly and safely transport critically injured soldiers to more sophisticated echelons of care. Exposure to the hypobaric conditions of the AE process may impose an additional physiologic risk on the TBI patient as well as a “second hit” inflammatory stimulus.
Conclusions
We review the known inflammatory effects of TBI and altitude exposure and propose that optimizing the post-traumatic inflammatory profile may assist in determining an ideal time to fly for head-injured soldiers.
Keywords: traumatic brain injury, aeromedical evacuation, inflammation, en route care, critical care aeromedic transport team
INTRODUCTION
Traumatic brain injury (TBI) and multiple organ dysfunction syndrome are leading causes of death in soldiers sustaining combat injuries [1]. While there have been significant recent advances in combat casualty care, especially with respect to initial resuscitation, TBI is still associated with mortality rates as high as 30% [2, 3], as well as pervasive morbidity [4, 5]. Of note, a significant number of TBI related deaths occur relatively late and are secondary to multiple organ failure and infectious complications, such as pneumonia [2, 4, 6].
An integral component of current combat casualty care doctrine is long-range aeromedical evacuation (AE). This system is designed to safely transport patients to appropriately advanced medical care facilities to receive needed treatment. The AE process has evolved, and in current practice, very seriously injured casualties are rapidly moved to increasing echelons of care, starting on the battlefield and ending in a tertiary medical center in the continental United States. Overall case fatality rate from Operation Iraqi Freedom (OIF) is lower than in prior conflicts, and the presumption has been made that the AE system has contributed to this improvement [7, 8]. However, the physiologic impact of AE on battle casualties has never been studied. Clear advantages of robust AE capability include the requirement for a smaller in-theater medical footprint and the ability to evacuate injured personnel to more sophisticated levels of care in a rapid fashion.
An initial insult such as a TBI may predispose patients to an exaggerated inflammatory response from a subsequent physiologic insult, or “second hit.” The nature of this “second hit” may vary and can potentially include hypotension, hypoxia, or infection [9]. There has been speculation that the “second hit” could be initiated or exacerbated by the AE process itself. The timing and environment of the AE process may impart physiologic stressors that adversely influence outcome. Unfortunately, there are few studies characterizing the systemic inflammatory response to AE following TBI, nor is there information available to guide optimization of the immunological status and minimize the inflammatory response to flight in an evacuated head-injured patient. The goal of this review is to examine our current knowledge of traumatic brain injury, inflammation, and the influence of AE.
TRAUMATIC BRAIN INJURY
TBI is defined as an acute nondegenerative, noncongenital injury to the head arising from blunt or penetrating trauma or from acceleration/deceleration forces [10]. More specifically, TBI is caused by a head injury that induces a decreased level of consciousness, amnesia, skull fracture, objective neurologic abnormality, intracranial lesion, or death [10]. TBI is graded as mild, moderate, or severe based on the level of consciousness as graded by the Glasgow coma scale (GCS) score following resuscitation [9]. TBI is the most common cause of death and disability in children and young adults. Each year in the United States, TBI affects 1.5 million people, resulting in 235,000 hospitalizations, 50,000 deaths, and permanent disability in 99,000 people [11, 12]. Patients who sustain even a moderate TBI can experience subsequent physical, behavioral, psychiatric, cognitive, and medical problems [5, 10]. The economic burden of traumatic brain injuries sustained in the United States in 2000 alone was an estimated $60 billion in total lifetime direct medical costs and indirect costs including lost productivity [13].
TBI IN CURRENT MILITARY CONFLICT
Given the exposure and vulnerability of the head and the violent nature of warfare, the risk of a full range of TBI in modern soldiers remains significant. The head, face, and neck comprise 12% of the total body surface area exposed during combat [3]. These areas remain vulnerable despite the evolution of protective body armor and helmets. The current Kevlar helmet is designed to protect against penetrating ballistic injury, but protects poorly against concussive injury [14]. The head and neck is the second most frequently injured region, with these injuries constituting the third leading cause of death from potentially survivable casualties thus far in OIF and Operation Enduring Freedom (OEF) [1, 15]. Of soldiers sustaining head, face, and neck injuries (HFNI), appropriate body armor, including a Kevlar helmet, is worn by 93% [3]. Over the first three and a half years of OIF/OEF, HFNI accounted for 30% to 40% of all reported injuries, and occur in up to 52% of all battle injuries and 22% of all non-battle injury patients (Fig. 1) [3, 16]. The head and neck region has sustained a higher proportion of blast injuries and fewer bullet wounds than any other body region. Blast injuries resulting from the increased use of explosive devices are estimated to result in TBI in nearly 60% of blast casualties [10]. This is a higher rate than any other military conflict of the 20th century (Fig. 2) [3, 5, 16, 17]. Multiple HFNI per patient are not uncommon, with isolated TBI responsible for 8% of all injuries [3, 16]. Of soldiers admitted to Walter Reed Army Medical Center following injury, 59% suffered some element of TBI, slightly more than half of which were classified as moderate to severe injury [18]. Thus, TBI represents a significant cause of death and disability in modern combat.
FIG. 1.
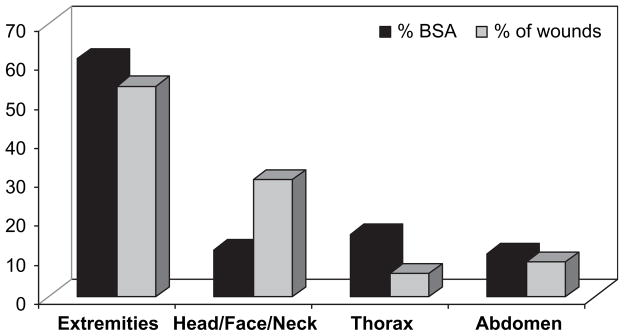
Wound distribution by region during OIF/OEF. %BSA=percent of body surface area (Adapted from [16]).
FIG. 2.

Percentage of head and neck wounds in each of the major conflicts of the 20th century. WWII=World War II; OIF/OEF=Operation Iraqi Freedom/Operation Enduring Freedom (Adapted from [16]).
SECONDARY BRAIN INJURY
Neurologic damage occurs both at the time of injury (primary TBI) and during evolution of the injury pattern over the next several days (secondary TBI). Secondary injury is the leading cause of inpatient deaths after TBI [9]. Most secondary injury is the result of progressive brain swelling from the accumulation of vasogenic edema. Increased cerebral edema is associated with elevated intracranial pressure and decreased cerebral perfusion, leading to superimposed tissue ischemia.
Patients with moderate to severe TBI are at significant risk of suffering secondary brain injury. The most common insults inducing secondary injury are early hypoxia and/or hypotension, which occur in 30% to 50% of head-injured patients in the pre-hospital setting [9, 19]. Patients with pre-hospital episodes of hypoxia, as defined by an arterial oxygen saturation less than 90%, or hypotension, as defined by a systolic blood pressure less than 90 mmHg, have as much as twice the risk of eventual death, suffer a greater degree of disability at discharge, and require longer hospital stays [9, 19, 20]. Strategies to decrease long-term sequelae of TBI must minimize secondary brain injury.
INFLAMMATORY RESPONSE TO TBI
The inflammatory response to TBI is increasingly acknowledged as an integral contributor to the severity and progression of primary and secondary head injury (reviewed by Morganti-Kossmann et al. [21] and Schmidt et al [22]). The cascade of inflammatory events, both localized and systemic, that occurs as a result of primary brain injury produces serious secondary damage that can double the mortality rate [23]. The brain has historically been regarded as an “immunologically privileged organ” due to its tight segregation from the peripheral circulation by the blood brain barrier (BBB) [22]. Primary brain injury, however, causes breakdown of the BBB, allowing the influx of systemic inflammatory mediators. In addition, the cerebral insult initiates a profound, prolonged endogenous neuroinflammatory response aimed at defending the intrathecal compartment from invading pathogens and repairing damaged brain tissue [22]. Once initiated, the immune functions of the resident cells of the central nervous system paradoxically contribute to the development of cerebral edema and exacerbation of tissue hypoxia through the release of pro-inflammatory cytokines, cytotoxic proteases, and reactive oxygen species (Fig. 3) [21, 22].
FIG. 3.

Contributors to development and progression of traumatic brain injury.
The hallmark of neuroinflammation in TBI is the functional duality of recruited immune cells, native glia, and secreted cytokines. Following TBI, no single factor, molecule, or cell acts in a purely detrimental or beneficial manner [21]. Cytokines and chemokines released as part of the systemic and neuroinflammatory responses to TBI contribute to the process of cellular damage induced by promoting the influx of inflammatory cells and mediators into the injured cerebral tissue. Conversely, cerebral inflammatory cytokines also play an integral role in the neuroprotective and neurotrophic processes responsible for the maintenance of continued neuronal function and initiation of wound healing.
Cytokines involved in mediating neuroinflammation are produced both systemically and intrathecally. These mediators stimulate the recruitment of inflammatory cells, regulate cerebrovascular permeability, and maintain activation of the resident neurons and supporting glia. In addition to playing an integral role in the propagation of the neuroinflammatory and neurotrophic responses to injury, cytokines are useful indicators of the presence of both organ-specific and systemic inflammation. Levels of several cytokines, including interleukins (IL)-1, -6, -8, -10, -18, tumor necrosis factor-alpha (TNFα), and transforming growth factor-beta, are elevated in both human and animal models of TBI [21, 22].
Elucidating the role of TNFα as an effector of the neural immune response to TBI has been the goal of several previous studies. TNFα encourages cerebral recruitment of systemic leukocytes, proteolytic degradation of the BBB, and inhibition of neuronal regeneration. Deficiency of TNFα is beneficial in the early posttraumatic period, but has been shown to be detrimental to long-term neurologic recovery, illustrating the dual role of inflammatory mediators in the propagation and resolution of brain injury [24, 25]. The timeline of TNFα expression is consistent among various rodent models of head injury, with upregulation by 1 h post-injury, maximal expression after 3–8 h, and a decline in release by 24 h. In addition, the type of TBI affects the focus of the TNFα response, with a detectable cerebral response in models of focal brain injury as compared to a systemic release following diffuse injury [21].
The IL-1 family of cytokines, including IL-1α, IL-1β, and IL-18, is well established as a promoter of neuroinflammation. Both the membrane-bound alpha and secreted beta forms of IL-1 have been shown to be upregulated within 1 h in both focal and diffuse models of TBI [22]. The deleterious effects of IL-1 are mediated by the IL-1 receptor, which is strongly expressed on microglia in focal brain injury and on neurons and astrocytes following a more diffuse brain injury. The neurotoxic effects of IL-1, including the up-regulation of matrix metalloproteases that contribute to BBB degradation, are enhanced by their synergistic activity with TNFα. Clinically, TBI patients with elevated cerebrospinal fluid (CSF) levels of IL-1 have worse neurologic outcomes [21]. Similarly, IL-18 is a potent activator of interferon-gamma (IFN-γ) and is elevated experimentally in brain tissue and clinically in CSF up to one week post brain injury [22]. Pharmacologic inhibition of IL-18 in murine models has been associated with improved neurologic recovery [26].
Interleukin-6 is widely recognized as a major mediator of the acute phase inflammatory response. Originally characterized as a neuroprotective cytokine and regulator of intracerebral homeostasis, IL-6 also asserts anti-inflammatory effects by inhibiting TNFα [21, 22]. However, intracerebral IL-6 also promotes neuroinflammation by upregulating local chemokine production and inducing the hepatic acute phase response after leaking into the peripheral circulation via the damaged BBB [22]. Animal models of TBI suggest that IL-6 is upregulated after 1 h in damaged brain tissue, with peak expression between 2 and 8 h post-injury. In humans, CSF levels of IL-6 are maximally increased between 3 and 6 d post-injury. Of all cytokines released into the CSF following TBI, IL-6 has the highest concentration. In human patients, elevated CSF IL-6 levels are associated with more favorable prognosis, whereas increased serum levels are prognostic of a worse outcome [27–30]. In addition, posttraumatic serum IL-6 levels have been shown to correlate with the degree of injury sustained, magnitude of systemic inflammation, and clinical outcome [31].
In contrast to the dual pro- and anti-inflammatory effects of the aforementioned cytokines after TBI, IL-10 appears to be strictly an anti-inflammatory cytokine that promotes immunosuppression [21]. IL-10 reduces neuroinflammation by inhibiting the pro-inflammatory roles of TNFα and IL-1. CSF levels of IL-10 are increased acutely within the first 24 h following TBI and correlate with decreases in TNFα. However, as IL-10 decreases local cerebral neuroinflammation, it also induces systemic immunosuppression in the polytrauma patient [21]. Although the direct effects of IL-10 may be anti-inflammatory, its promotion of peripheral immunosuppression may indirectly contribute to secondary brain injury by increasing susceptibility to infection. Thus, IL-10 demonstrates an important immunologic conundrum in the post-TBI patient: increased intracranial cytokine release may improve the local neurologic outcome but, paradoxically, worsen the ultimate outcome for the patient.
Neuroinflammation after TBI involves the release of chemokines in addition to the production of cytokines. Two chemokines integral to the recruitment of peripheral blood leukocytes to sites of local injury are IL-8 and monocyte chemoattractant protein-1 (MCP-1). IL-8 is a potent chemotactic factor for neutrophils that contributes to the secondary brain injury induced by neuroinflammation by promoting protease release and BBB breakdown. Increased CSF levels of IL-8 have been demonstrated in patients within 6 h of TBI. MCP-1 is essential for peripheral monocyte recruitment and is elevated in the damaged brain between 4 and 16 h post-injury, corresponding to the perivascular appearance of monocytes/macrophages on cerebral immunohistochemistry after TBI [21]. In addition, contused human brain tissue contains high levels of MCP-1 [32].
Taken together, data examining the presence and function of cytokines and chemokines in TBI allow us to begin to understand the important role of the acute and chronic inflammatory response in this condition. Attenuation of the inflammatory response may allow therapeutic intervention in patients suffering TBI.
RELATIONSHIP OF ALTITUDE AND INFLAMMATION
Exposure to moderate altitude change occurs regularly during commercial (4000 to 6000 ft) as well as military AE (4500 to 8800 ft) flights. Passenger cabins are routinely pressurized to protect occupants from hypobaric conditions at flight altitudes. At cabin pressures equivalent to 5000 to 8000 feet, passengers without medical problems regularly have a peripheral oxygen saturation level of 89% [33]. In patients with poor perfusion, oxygen saturation levels may decrease into a more precarious range of the hemoglobin-oxygen saturation curve [34]. However, increased cabin pressurization reduces fuel efficiency, increases flight duration, decreases energy available for other aircraft systems, limits the operational lifetime of aluminum airframes, and necessitates increased structural weight, all of which are counterproductive in military settings [35].
Unacclimatized travelers to terrestrial elevations above 6500 ft may experience a syndrome known as acute mountain sickness (AMS). AMS is characterized by headache, anorexia, nausea, emesis, fatigue, and sleep disturbance [36]. Onset of symptoms may occur within 6 h of elevation change. With continued high altitude exposure, abnormal respiratory or neurologic signs and symptoms of AMS can progress to high altitude pulmonary edema (HAPE) or high altitude cerebral edema (HACE). HAPE consists of a spectrum of pulmonary dysfunction, ranging from minimal dyspnea on exertion to a productive cough with blood-tinged sputum. HACE often accompanies HAPE and is characterized by the development of ataxia and altered consciousness. The incidence and severity of AMS, HAPE, and HACE depend on the altitude achieved, rate of ascent to altitude, and individual susceptibility. Additional risk factors may include concomitant respiratory tract infection, dehydration, and age greater than 50 years [36]. Of note, each of these risk factors may be present in acutely injured patients with TBI, potentially placing them at higher risk for altitude-related illness during AE.
The exact mechanisms causing syndromes of high altitude illness are currently unknown. A common hypothesis for the etiology of AMS, HAPE, and HACE is hypobaric hypoxia. Hypoxemia is theorized to elicit hemodynamic and neurohumoral responses that are responsible for altering the permeability of the BBB, increasing cerebral blood flow, and reducing the formation of vasodilatory nitric oxide in the pulmonary endothelium. These changes produce the cerebral vasogenic edema and exaggerated pulmonary hypertension thought to contribute to the development of HACE and HAPE, respectively. In addition, the severity of high altitude illness symptoms is inversely related to the arterial oxygen content [35]. Moreover, hypoxia induces the release of IL-1β, IL-6, and TNFα in rodents and IL-6 alone in humans [37, 38]. Thus, altitude-induced hypoxemia may be responsible for generating the symptoms associated with high-altitude illness and a concomitant inflammatory response.
Recent evidence suggests that preexisting inflammation may predispose an individual to acute high altitude illness. Durmowicz et al. showed that the presence of established inflammation-producing respiratory illnesses contribute to the development of HAPE in children arriving at higher terrestrial elevations [39]. Other investigators further demonstrated that endotoxin priming prior to high altitude exposure may cause increased vascular permeability and pulmonary edema via neutrophil activation [40]. This pattern is consistent with a “two hit” model of critical illness, whereby a single insult primes the body for a much more severe response to a secondary insult [41, 42]. There is significant concern that TBI may act as an initial insult that primes the body for a second hit from altitude-induced inflammatory changes incurred by post-injury AE. In this setting, AE may worsen severity of injury, morbidity, and mortality from TBI.
AEROMEDICAL EVACUATION
Over the course of 140 y, establishment of an AE system has become an integral component of military patient movement in peacetime and while at war. Although the history of AE dates to the airlift of wounded via hydrogen-filled balloons in 1870 during the Franco-Prussian War, AE saw limited implementation until World War II. The modern US AE system has evolved into a worldwide patient airlift capable of providing rapid and safe transport of military patients to specialized medical facilities (Fig. 4) [43]. Treatment, stabilization, and evacuation of casualties to higher echelons of care are fundamental components of military medical doctrine that preserve deployed medical resources for further casualty reception [44].
FIG. 4.

Time course of casualty treatment and evacuation through echelons of military medical care. (Color version of figure is available online.)
The introduction of the Air Force Critical Care Aeromedic Transport Team (CCATT) has been integral to the ability to transport critically ill patients from austere far forward battlefield environments to sophisticated international and domestic military medical institutions (Fig. 4). En route care, as provided by CCAT teams during OIF/OEF, has effectively and safely enabled transport of injured soldiers to higher echelon hospitals more rapidly than in any previous conflict [45, 46]. During OIF/OEF, the average time from injury to hospitalization in the continental United States has been 4 to 5 d. This represents a sharp reduction from 45 d during the Vietnam conflict [8, 47]. Over the 18-mo period from January 2007 to June 2008, 12% of CCATT transported patients had a diagnosis of TBI. Of these head-injured patients, 80% were mechanically ventilated, and 35% had ICP monitors throughout transport (CCATT Database, December 2008). In current military medicine, CCATT-assisted advanced care of the mobile patient has become essential to transporting acutely injured and critically ill casualties.
Coordinated AE is integral to the existing military trauma system, which has reduced mortality from battlefield wounds to the lowest achieved in the history of American wars [7, 8]. While AE is an important component of this system, the current improved survival rate may not be attributed to the rapid movement of patients out of the combat theater. There is concern that AE itself, in certain circumstances, may propagate the damage initiated by a traumatic injury. Up to 7% of passengers on long commercial flights experience symptoms similar to those of AMS after as few as 3 h. Because barometric pressures in the aircraft cabins are similar to those at terrestrial altitudes at which AMS occurs, it is possible that some of these symptoms are related to decreased arterial oxygen saturation and are manifestations of AMS [35]. Of significance to the evacuated combat casualty, altitude-related concerns are most imposing closer to the time of injury or illness [34]. AE exposes patients to additional physiologic stresses, including hypobaric hypoxia at lower cabin pressurization, increased gas expansion in susceptible cavities, variation in temperature, noise, vibration, decreased humidity, fatigue, and the effects of acceleration during rapid tactical ascent [49]. A change from sea level to an altitude of 8000 ft expands the volume of trapped gas by approximately 35%, putting those with head and chest trauma or recent surgery at greatest risk [34, 50, 51]. These stresses, though generally well tolerated by healthy individuals, may cause the deterioration of critically injured patients despite advances in aeromedic transport care. Within 48 h of AE, 48% of patients undergo additional surgery, suggesting that combat injuries sustained are still in evolution and therefore susceptible to physiologic insult during AE [48].
AE AND TBI
The goal of treatment during the acute phase of TBI is to prevent secondary brain injury while rapidly transporting the patient to definitive care [23]. Casualties evacuated by airlift may be relatively susceptible to hypoxia due to the lower partial pressure of oxygen at altitude. Aeromedical transport of casualties with TBI may place them at increased risk for secondary brain injury from tissue hypoxia. Hypoxemia following TBI is associated with worse neurologic outcome and increased mortality in patients with moderate or severe TBI [52].
Multi-organ failure induced by systemic inflammation remains the most frequent cause of late death in trauma patients who survive initial resuscitation and stabilization [53]. Outcomes following TBI are strongly related to the post-traumatic occurrence of non-neurologic organ dysfunction, with up to 89% of patients with severe TBI developing dysfunction of at least one non-neurologic organ, and 35% progressing to organ failure [2]. Post-TBI inflammation and infection are the primary contributors to the initiation of organ dysfunction [4]. Thus, the systemic and cerebral inflammatory responses to TBI may prime the patient for an exaggerated inflammatory response to altitude exposure and hypobaric hypoxemia, thereby creating a “two hit” injury paradigm that places acutely evacuated TBI patients at additional risk for multi-organ dysfunction. Unfortunately, there is a paucity of data available about the effects of AE on the progression and outcome of TBI in the peri-flight period. We theorize that serial assessments of the head-injured patient’s serum inflammatory cytokine profile may help to optimize post-injury time to fly by minimizing altitude-induced exacerbation of the post-TBI neuroinflammatory response. Air transport after resolution or reduction of the initial systemic inflammatory response to head injury may reduce the head-injured casualty’s vulnerability to the detrimental immune effects of AE.
CONCLUSIONS
In summary, TBI has become the signature injury of current military conflict. Head injuries are responsible for a significant portion of combat-related mortality and will contribute to ongoing morbidity and disability in injured soldiers for years to come. Recent research has defined a role for inflammation, both local and systemic, following head injury. This inflammation, along with post-traumatic hypotension and hypoxia, places head-injured patients at considerable risk for secondary brain injury and worse neurologic outcomes. The physiologic insults inherent in AE may impart and compound these same risks in the evacuated head-injured patient. In order to optimize timing of aeromedic evacuation and minimize the “second hit” induced by the process of evacuation, further studies are needed to characterize the systemic inflammatory response to AE, accurately delineate the post-TBI systemic and neuroinflammatory profiles, and measure the additional immunological impact of post-TBI AE at various times following injury. By identifying an “ideal time to fly” based on inflammatory response to injury and AE, the potential secondary injury induced by the AE experience may be reduced, leading to improved TBI outcomes from battlefield injuries.
Acknowledgments
This work was performed at the University of Cincinnati, Cincinnati, Ohio.
The authors acknowledge support in part for this study by the United States Air Force/Henry Jackson Foundation award FA8650-05-2-6518.
References
- 1.Kelly JF, Ritenour AE, McLaughlin DF, et al. Injury severity and causes of death from Operation Iraqi Freedom and Operation Enduring Freedom: 2003–2004 versus 2006. J Trauma. 2008;64:S21. doi: 10.1097/TA.0b013e318160b9fb. [DOI] [PubMed] [Google Scholar]
- 2.Zygun DA, Kortbeek JB, Fick GH, et al. Non-neurologic organ dysfunction in severe traumatic brain injury. Crit Care Med. 2005;33:654. doi: 10.1097/01.ccm.0000155911.01844.54. [DOI] [PubMed] [Google Scholar]
- 3.Wade AL, Dye JL, Mohrle CR, et al. Head, face, and neck injuries during Operation Iraqi Freedom II: Results from the US Navy-Marine Corps Combat Trauma Registry. J Trauma. 2007;63:836. doi: 10.1097/01.ta.0000251453.54663.66. [DOI] [PubMed] [Google Scholar]
- 4.Berthiaume L, Zygun D. Non-neurologic organ dysfunction in acute brain injury. Crit Care Clin. 2007;22:753. doi: 10.1016/j.ccc.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Okie S. Traumatic brain injury in the war zone. N Engl J Med. 2005;352:2043. doi: 10.1056/NEJMp058102. [DOI] [PubMed] [Google Scholar]
- 6.Pilitsis JG, Rengachary SS. Complications of head injury. Neurol Res. 2001;23:227. doi: 10.1179/016164101101198389. [DOI] [PubMed] [Google Scholar]
- 7.Carlton PK, Jenkins DH. The mobile patient. Crit Care Med. 2008;36:S255. doi: 10.1097/CCM.0b013e31817da609. [DOI] [PubMed] [Google Scholar]
- 8.Gawande A. Casualties of war–military care for the wounded from Iraq and Afghanistan. N Engl J Med. 2004;351:2471. doi: 10.1056/NEJMp048317. [DOI] [PubMed] [Google Scholar]
- 9.Ghajar J. Traumatic brain injury. Lancet. 2000;356:923. [Google Scholar]
- 10.Galarneau MR, Woodruff SI, Dye JL, et al. Traumatic brain injury during Operation Iraqi Freedom: Findings from the United States Navy–Marine Corps Combat Trauma Registry. J Neurosurg. 2008;108:950. doi: 10.3171/JNS/2008/108/5/0950. [DOI] [PubMed] [Google Scholar]
- 11.A Report to Congress. Vol. 2004. Centers for Disease Control; 2001. Traumatic Brain Injury in the United States. [Google Scholar]
- 12.Langlois JA, Rutland-Brown W, Thomas KE. Traumatic brain injury in the United States: Emergency department visits, hospitalizations, and deaths. Atlanta: Centers for Disease Control and Prevention, Nation Center for Injury Prevention and Control; 2006. Available at: www.cdc.gov/ncipc/factsheets/tbi.htm. [Google Scholar]
- 13.Finkelstein E, Corso P, Miller T, et al. The incidence and economic burden of injuries in the United States. New York: Oxford University Press; 2006. [Google Scholar]
- 14.Sarron JC, Dannawi M, Faure A, et al. Dynamic effects of a 9mm missile on cadaveric skull protected by aramid, polyethylene, or aluminum plate: An experimental study. J Trauma. 2004;57:236. doi: 10.1097/01.ta.0000133575.48065.3f. [DOI] [PubMed] [Google Scholar]
- 15.Murray CK. Epidemiology of infections associated with combat-related injuries in Iraq and Afghanistan. J Trauma. 2008;64:S232. doi: 10.1097/TA.0b013e318163c3f5. [DOI] [PubMed] [Google Scholar]
- 16.Owens BD, Kragh JF, Wenke JC, et al. Combat wounds in Operation Iraqi Freedom and Operation Enduring Freedom. J Trauma. 2008;64:295. doi: 10.1097/TA.0b013e318163b875. [DOI] [PubMed] [Google Scholar]
- 17.Rustemeyer J, Kranz V, Bremerich A. Injuries in combat 1982–2005 with particular reference to those to the head and neck: A review. Br J Oral Maxillo Surg. 2007;45:556. doi: 10.1016/j.bjoms.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Warden D. Military TBI during the Iraq and Afghanistan wars. J Head Trauma Rehabil. 2006;21:398. doi: 10.1097/00001199-200609000-00004. [DOI] [PubMed] [Google Scholar]
- 19.Chestnut RM, Marshall LF, Klauber MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34:216. doi: 10.1097/00005373-199302000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Chi JH, Knudson MM, Vassar MJ, et al. Prehospital hypoxia affects outcome in patients with traumatic brain injury: A prospective multicenter study. J Trauma. 2006;61:1134. doi: 10.1097/01.ta.0000196644.64653.d8. [DOI] [PubMed] [Google Scholar]
- 21.Morganti-Kossmann MC, Satgunaseelan L, Bye N, et al. Modulation of immune response to head injury. Injury. 2007;38:1392. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt OI, Heyde CE, Ertel W, et al. Closed head injury - an inflammatory disease? Brain Res Rev. 2005;48:388. doi: 10.1016/j.brainresrev.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 23.Morris M. Transport considerations for the head-injured patient: Are we contributing to secondary injury? J Air Med Transp. 1992;11:9. doi: 10.1016/s1046-9095(05)80391-5. [DOI] [PubMed] [Google Scholar]
- 24.Scherbel U, Raghupathi R, Nakamura M, et al. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Nat Acad Sci. 1999;96:8721. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stahel PF, Shohami E, Younis FM, et al. Experimental closed head injury: An analysis of neurologic outcome, blood-brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for proinflammatory cytokines. J Cereb Blood Flow Metab. 2000;20:369. doi: 10.1097/00004647-200002000-00019. [DOI] [PubMed] [Google Scholar]
- 26.Yatsiv I, Morganti-Kossmann MC, Perez D, et al. Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J Cereb Blood Flow Metab. 2002;22:971. doi: 10.1097/00004647-200208000-00008. [DOI] [PubMed] [Google Scholar]
- 27.Arand M, Melzner H, Kinzl L, et al. Early inflammatory mediator response following isolated traumatic brain injury and other major trauma in humans. Langenbecks Arch Surg. 2001;386:241. doi: 10.1007/s004230100204. [DOI] [PubMed] [Google Scholar]
- 28.Singhal A, Baker AJ, Hare GM, et al. Association between cerebrospinal fluid interleukin-6 concentrations and outcome after severe human traumatic brain injury. J Neurotrauma. 2002;19:929. doi: 10.1089/089771502320317087. [DOI] [PubMed] [Google Scholar]
- 29.Winter CD, Pringle AK, et al. Raised parenchymal interleukin-6 levels correlate with improved outcome after traumatic brain injury. Brain. 2004;127:315. doi: 10.1093/brain/awh039. [DOI] [PubMed] [Google Scholar]
- 30.Kushi H, Saito T, Makino K, et al. IL-8 is a key mediator of neuroinflammation in severe traumatic brain injuries. Acta Neurochir Suppl. 2003;86:347. doi: 10.1007/978-3-7091-0651-8_74. [DOI] [PubMed] [Google Scholar]
- 31.Giannoudis PV. Current concepts of the inflammatory response after major trauma: An update. Int J Care Inj. 2003;34:397. doi: 10.1016/s0020-1383(02)00416-3. [DOI] [PubMed] [Google Scholar]
- 32.Stefini R, Catenacci E, Piva S, et al. Chemokine detection in the cerebral tissue of patients with posttraumatic brain contusions. J Neurosurg. 2008;108:958. doi: 10.3171/JNS/2008/108/5/0958. [DOI] [PubMed] [Google Scholar]
- 33.Cottrell JJ, Lebovitz BL, Fennell RG, et al. In-flight arterial saturation: Continuous monitoring by pulse oximetry. Aviat Space Environ Med. 1995;66:126. [PubMed] [Google Scholar]
- 34.Teichman PG, Donchin Y, Kot RJ. International aeromedic evacuation. N Eng J Med. 2007;356:262. doi: 10.1056/NEJMra063651. [DOI] [PubMed] [Google Scholar]
- 35.Muhm JM, Rock PB, McMullin DL, et al. Effect of aircraft-cabin altitude on passenger discomfort. N Eng J Med. 2007;357:18. doi: 10.1056/NEJMoa062770. [DOI] [PubMed] [Google Scholar]
- 36.Basnyat B, Murdoch DR. High altitude illness. Lancet. 2003;361:1967. doi: 10.1016/S0140-6736(03)13591-X. [DOI] [PubMed] [Google Scholar]
- 37.Ertel W, Morrison MH, Ayala A, et al. Hypoxemia in the absence of blood loss or significant hypotension causes inflammatory cytokine release. Am J Physiol. 1995;269:R160. doi: 10.1152/ajpregu.1995.269.1.R160. [DOI] [PubMed] [Google Scholar]
- 38.Klausen T, Olsen NV, Poulsen TD, et al. Hypoxemia increases serum interleukin-6 in humans. Eur J Appl Physiol Occup Physiol. 1997;76:480. doi: 10.1007/s004210050278. [DOI] [PubMed] [Google Scholar]
- 39.Durmowicz AG, Noordeweir E, Nicholas R, et al. Inflammatory processes may predispose children to high-altitude pulmonary edema. J Pediatr. 1997;130:838. doi: 10.1016/s0022-3476(97)80033-9. [DOI] [PubMed] [Google Scholar]
- 40.Ono S, Westcott JY, Chang SW, et al. Endotoxin priming followed by high altitude causes pulmonary edema in rats. J Appl Physiol. 1993;74:1534. doi: 10.1152/jappl.1993.74.4.1534. [DOI] [PubMed] [Google Scholar]
- 41.Grocott M, Montgomery H, Vercueil A. High-altitude physiology and pathophysiology: Implications and relevance for intensive care medicine. Crit Care. 2007;11:203. doi: 10.1186/cc5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am. 1995;75:257. doi: 10.1016/s0039-6109(16)46587-4. [DOI] [PubMed] [Google Scholar]
- 43.Merwin CA. US Air Force patient airlift. J Air Med Transp. 1990;9:18. doi: 10.1016/s1046-9095(05)80527-6. [DOI] [PubMed] [Google Scholar]
- 44.Harman DR, Hooper TI, Gackstetter GD. Aeromedic evacuations from Operation Iraqi Freedom: A descriptive study. Mil Med. 2005;170:521. doi: 10.7205/milmed.170.6.521. [DOI] [PubMed] [Google Scholar]
- 45.Johannigman JA. Critical care aeromedic teams (CCATT): Then, now and what’s next. J Trauma. 2007;62:S35. doi: 10.1097/TA.0b013e31806540f3. [DOI] [PubMed] [Google Scholar]
- 46.Johannigman JA. Maintaining the continuum of en route care. Crit Care Med. 2008;36:S377. doi: 10.1097/CCM.0b013e31817e31e1. [DOI] [PubMed] [Google Scholar]
- 47.Richardson MW. Casualty evacuations: Transport of the severely injured. J Trauma. 2007;62:S64. doi: 10.1097/TA.0b013e318065adf3. [DOI] [PubMed] [Google Scholar]
- 48.Montgomery SP, Swiecki CW, Shriver CD. The evaluation of casualties from Operation Iraqi Freedom on return to the continental United States from March to June 2003. J Am Coll Surg. 2005;201:7. doi: 10.1016/j.jamcollsurg.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 49.Turkan H, Sener S, Tugcu H, et al. Considerations in the aeromedic evacuation of the critically ill blast victim: Lessons learned. Mil Med. 2006;171:586. doi: 10.7205/milmed.171.7.586. [DOI] [PubMed] [Google Scholar]
- 50.Andersson N, Grip H, Lindvall P, et al. Air transport of patients with intracranial air: Computer model of pressure effects. Aviat Space Environ Med. 2003;74:138. [PubMed] [Google Scholar]
- 51.Helling E, McKinlay AJ. Considerations for the head-injured air-evacuated patient: A case report of frontal sinus fracture and review of the literature. Mil Med. 2005;170:577. doi: 10.7205/milmed.170.7.577. [DOI] [PubMed] [Google Scholar]
- 52.Grissom CK, Weaver LK, Clemmer TP, et al. Theoretical advantage of oxygen treatment for combat casualties during medical evacuation at high altitude. J Trauma. 2006;61:461. doi: 10.1097/01.ta.0000221699.71596.9d. [DOI] [PubMed] [Google Scholar]
- 53.Surbatovic M, Filipovic N, Radakovic S, et al. Immune cytokine response in combat casualties: Blast or explosive trauma with or without secondary sepsis. Mil Med. 2007;172:190. doi: 10.7205/milmed.172.2.190. [DOI] [PubMed] [Google Scholar]