

Abstract
Activity-dependent stimuli induced a calcineurin-mediated dephosphorylation of the transcriptional factor MEF2A at serine408 and promoted a switch from SUMOylation to acetylation at lysine403 which led to MEF2A transcriptional activation. We previously identified SENP2 is the de-SUMOylation enzyme for MEF2A and promotes MEF2A-dependent transcription. We report here a requirement for APCCdh1-SENP2-MEF2A axis in the regulation of MEF2A transcriptional activation. APCCdh1 interacts with and targets SENP2 for ubiquitination and destruction in the cytoplasm by recognizing a conserved canonical D-box motif in SENP2. Moreover, Cdh1 regulates the transcriptional activity of MEF2A in a SENP2 dependent manner. Activity-dependent stimuli prevented APCCdh1-induced SENP2 ubiquitination, promoted SENP2 nuclear accumulations, and caused MEF2A de-SUMOylation and MEF2A acetylation, leading to MEF2A transcriptional activation. Thus, our findings defined a post-transcriptional mechanism underlying activity-dependent stimuli-induced MEF2A transcriptional activation.
Keywords: activity-dependent stimuli, APC-Cdh1, MEF2A, SENP2
Introduction
Activity-dependent stimuli can modulate neuronal plasticity, which is a result of the brain's ability to convert transient stimuli into long-lasting alterations in neuronal structure and function. This results in an increase (potentiation) or a decrease (depression) in synaptic strength.1 This process involves changes in receptor trafficking, protein–protein interactions, local mRNA translation, new gene synthesis and protein post-translational modifications including phosphorylation, acetylation, ubiquitination and SUMOylation.2-5
SUMOylation is the covalent linkage of a small ubiquitin-related modifier (SUMO) to the ε-amine of lysine residues in target proteins. It is an important regulatory mechanism for modifying protein activity, stability, and cellular localization.6 Mammalian cells express 4 SUMO paralogs: SUMO1–SUMO4. Human SUMO2 and SUMO3 are about 95% identical to each other, whereas they share only 45% identity with SUMO1. SUMO4 is the least well characterized isoform and is expressed mainly in the kidney, lymph node and spleen, whereas SUMO1, SUMO2 and SUMO3 are ubiquitously expressed.7 SUMOylation is a high dynamic process that is catalyzed by the activating (E1), conjugating (E2) and ligating (E3) enzymes and reversed by a family of Sentrin/SUMO-specific proteases (SENPs).8 In mammalian cells, 6 SENPs have been identified, which have different substrate specificity and subcellular localization. SENPs are divided into 3 subfamilies on the basis of their sequence homology, cellular location and substrate specificity.9 The first subfamily includes SENP1 and SENP2, which have broad substrate specificity.10,11 SENP2 is a nucleocytoplasmic shuttling protease that appears to have a activity similar to that of SENP1 when overexpressed.12 Loss of mouse SENP2 caused embryonic lethality.13 SENP2 is essential for embryonic cardiac development through regulation of the SUMOylation status of Pc2/CBX4 and has a specific role in the G-S transition, which is required for mitotic and endoreduplication cell cycles in trophoblast proliferation and differentiation.13,14 The second subfamily includes SENP3 and SENP5, both of which are nucleolar proteins with preference for SUMO-2/3.15 The third subfamily includes SENP6 and SENP7, which have an extra loop in their catalytic domains.16
The MEF2A transcriptional factor is highly expressed in the brain and plays a key role in synaptic dendritic development in the cerebellar cortex.17 The modifications of MEF2A required for postsynaptic differentiation occur within a phosphorylation-regulated SUMOylation-acetylation switch (SAS) peptide motif.18 Activity-dependent stimuli increased MEF2A transcriptional activity by activating a highly programmed post-transcriptional event taken place on MEF2A, including MEF2A de-phosphorylation, MEF2A de-SUMOylation and MEF2A acetylation.19 Calcineurin dephosphorylates MEF2A Ser408 which resulted in Lys403 de-SUMOylation, sequential Lys403 acetylation and de-represses MEF2A-induced transcription.18 Further study discovered that PIASx promoted postsynaptic dendritic morphogenesis by enhancing the SUMOylation of MEF2A.20 We previously identified SENP2 was the predominant de-SUMOylation enzyme for MEF2A by using unbiased functional loss of function screen.21 We found that SENP2 regulates the transcriptional function of MEF2A via direct de-SUMOylation. In conformity with these clues, MEF2A was heavily SUMOylated in SENP2−/− embryo.21 However, how endogenous SENP2 is regulated during this process is still undefined.
Results
Activity-dependent stimuli prevent SENP2 ubiquitination and degradation
We first investigated whether endogenous SENP2 was regulated under KCl-induced depolarization manipulation by using primary rat cortex neuron cells. KCl-induced depolarization promote the transcriptional activity of MEF2A has been well documented.18,19 Indeed, KCl treatment significantly enhanced MEF2A transcriptional activation as evidenced by the accumulation of SynGAP, one of the downstream target genes of MEF2A18 (Fig. 1A). Under the same circumstances, endogenous SENP2 protein was also accumulated in a time-dependent manner (Fig. 1A). However, the mRNA level of SENP2 was almost unchanged, suggesting a post-transcriptional regulation was taking place (Fig. S1). SENP2 has been reported to be a short-lived protein and destructed via ubiquitin-proteasome system.12 We proposed that KCl-induced endogenous SENP2 accumulation could be due to impaired SENP2 destruction. Indeed, the half-life of endogenous SENP2 was significantly increased in KCl-treated SHSY5Y cells (Fig. 1B). Moreover, KCl stimuli dramatically decreased the ubiquitination form of SENP2 (Fig. 1C). Consistent with these data, KCl stimuli further enhanced the de-SUMOylation activity of SENP2 toward MEF2A and caused MEF2A SAS transition (Fig. 1D) and SENP2-mediated MEF2A transcriptional activation (Fig. S2). Furthermore, silencing the expression of SENP2 by siRNA largely abolished KCl-induced MEF2A activation (Fig. 1E, F). Taken together, these data indicated that activity-dependent stimuli prevents SENP2 ubiquitination and degradation accompanied with MEF2A activation.
Figure 1.

Activity-dependent stimuli activate MEF2A activation by preventing SENP2 ubiquitination and degradation. (A) Lysates of primary rat cortex neuron cells depolarized with 50 mM KCl for the indicated time were immunoblotted for SynGAP and SENP2. β-actin was used as loading control. The number on the bottom of Western blot lanes indicated signal intensity of SENP2 or SynGAP protein against β-actin. The signal intensity of SENP2 or SynGAP protein against β-actin at 0 min was set as 1. (B) SHSY5Y cells pretreated with 50 mM KCl or 150 mM NaCl (corresponds to physiological conditions: 0.9% NaCl) for 1 h and 20 mM CHX was added for the indicated time. Lysates from these cells were immunoblotted for SENP2. Histograms represent the ratio of the signal intensity of SENP2 protein quantified against β-actin. (C) SHSY5Y cells were transfected with Flag-SENP2 and HA-ubiquitin plasmids for 36 h. Cells were then treated with 50 mM KCl or 150 mM NaCl for 2 h. Cells were lysed and subjected to immunoprecipitation with Flag M2 beads. Immunoprecipitated proteins were immunoblotted with anti-HA antibody. (D) SHSY5Y cells were transfected with indicated plasmids for 36 h. Cells were then treated with 50 mM KCl or 150 mM NaCl for 2 h. Cells were lysed and subjected to immunoprecipitation with Flag M2 beads. Immunoprecipitated proteins were immunoblotted with anti-HA antibody. (E) SHSY5Y cells were transfected with non-specific siRNA or siRNA against SENP2 for 48 h. Cells were then treated with 50 mM KCl or 150 mM NaCl for 2 h. The whole cell lysates were immunoblotted for SENP2. (F) SHSY5Y cells were transfected with MEF2A and MEF-luciferase reporter plasmids with non-specific siRNA or siRNA against SENP2 for 36 h. Cells were then treated with 50 mM KCl or 150 mM NaCl for 2 h, the luciferase activity was measured. Transfection efficiency was normalized by Renilla luciferase expression, and the results are presented as activation over that for non-specific siRNA transfected group. The y axis represents normalized luciferase activity ± SEM (n = 3). **represents P < 0.01, *** represents P < 0.001.
Activity-dependent stimuli promote SENP2 nuclear translocation
SENP2 is reported to be a nucleocytoplasmic shuttling protein and is ubiquitinated in the cytoplasm.12 We then asked that whether activity-dependent stimuli promoted SENP2 nuclear translocation. To test this possibility, primary rat cortex neuron cells were treated with 50 mM KCl to induce SENP2 accumulation. The nuclear-cytoplasmic fraction of primary rat cortex neuron cells was conducted. As depicted in Fig. 2A, KCl stimuli significantly promoted SENP2 nuclear translocation. There is a bipartite nuclear localization signal (NLS) and a CRM1-dependent nuclear export signal (NES) in SENP212 (Fig. 2B). A cytoplasm-localized SENP2 made by introducing mutations in the NLS (SENP2 mNLS) was unable to accumulate in response to KCL stimuli in SHSY5Y cells (Fig. 2C, Fig. S3). KCl stimuli could also not decrease the ubiquitination form of SENP2 mNLS (Fig. S4). Moreover, overexpression of SENP2 mNLS could not increase the transcriptional activity of MEF2A when compared with SENP2 WT (Fig. 2D). leptomycin B, an inhibitor of CRM1, could specifically inhibit the CRM1-dependent nuclear export activity of the NES.22 LMB treatment did not affect the mRNA level of SENP2 (Fig. S5). However, LMB treatment inhibited the nuclear export of SENP2 (Fig. 2E), prevented SENP2 ubiquitination (Fig. 2F), promoted MEF2A de-SUMOylation (Fig. 2G), transcriptional activation (Fig. 2H) and downstream target gene SynGAP expression (Fig. 2I). Conclusively, these data suggested that activity-dependent stimuli prevent SENP2 ubiquitination and degradation by promoting its nuclear translocation.
Figure 2.

Activity-dependent stimuli promote SENP2 nuclear translocation. (A) SHSY5Y cells were treated with 50 mM KCl or 150 mM NaCl for 2 h and the nuclear-cytoplasmic fraction of these cells was conducted. Lysates from each fraction were immunoblotted for SENP2. Tubulin and Lamin B were used to indicate cytoplasmic or nuclear fraction, respectively. The number on the bottom of Western blot lanes indicated signal intensity of SENP2 protein against β-Tubulin or Lamin B. The signal intensity of SENP2 protein against β-Tubulin or Lamin B without KCl treatment was set as 1. (B) Schematic diagrams of the NLS, NES and Protease domain of SENP2. (C) SHSY5Y cells were transfected with Flag-SENP2 WT or Flag-SENP2 mNLS for 36 h. Cells were treated with 50 mM KCl or 150 mM NaCl for 2 h. The whole cell lysates were immunoblotted with Flag antibody. (D) SHSY5Y cells were transfected with indicated plasmids for 36 h. Cells were then treated with 50 mM KCl or 150 mM NaCl for 2 h, the luciferase activity was measured. The y axis represents normalized luciferase activity ± SEM (n = 3). **represents P < 0.01. (E) SHSY5Y cells were transfected with Flag-SENP2 for 32 h and treated with 10 nM LMB or DMSO for 2 h and the nuclear-cytoplasmic fraction of these cells was conducted. Lysates from each fraction were immunoblotted for Flag-SENP2. Tubulin and Lamin B were used to indicate cytoplasmic or nuclear fraction, respectively. (F) SHSY5Y cells were transfected with Flag-SENP2 and His-ubiquitin plasmids for 36 h. Cells were then treated with 10 nM LMB. Cells were lysed and subjected to immunoprecipitation with Flag M2 beads. Immunoprecipitated proteins were immunoblotted with anti-His antibody. (G) SHSY5Y cells were transfected with Flag-MEF2A and HA-SUMO1 for 36 h. Cells were then treated with 10 nM LMB for 2 h. Cells were lysed and subjected to immunoprecipitation with Flag M2 beads. Immunoprecipitated proteins were immunoblotted with anti-HA and Flag antibodies. (H) SHSY5Y cells were transfected with MEF2A and MEF-luciferase reporter plasmids for 36 h. Cells were then treated with 10 nM LMB for 2 h, the luciferase activity was measured. The y axis represents normalized luciferase activity ± SEM (n = 3). **represents P < 0.01. (I) Lysates of primary rat cortex neuron cells treated with 10 nM LMB or DMSO were immunoblotted for SynGAP and SENP2.
SENP2 contains a canonical D-box motif
To identify which E3 ligase is required for the degradation of SENP2, we first checked the protein sequence of SENP2 for potential degrons. We noted that SENP2 contains a canonical ‘destruction box’ (D-box) motif (R-X-X-L-X-X-X-X-N/D/E) (Fig. 3A). D-box and KEN are 2 degradation motifs have been identified in the substrates of Anaphase-Promoting Complex (APC), which is a multi-subunit E3 protein.23 APC is responsible for ubiquitination of cell cycle regulators in non-neuronal cells but also highly expressed and has additional function in the post-mitotic neurons.24 Most APC substrates have D box and some have either a KEN box or both D and KEN boxes. In some cases 2 D boxes appear in the same protein. It is worth to mention that, this D-box motif of SENP2 is evolutionarily conserved among different species including Mus musculus, Rattus norvegicus, Pongo abelii and Homo sapiens (Fig. 3B). Tosyl-L-arginine methyl ester (TAME), which is a potent inhibitor of APC, could directly bind to APC and disrupts the interactions between APC and its activator proteins Cdc20 or Cdh1.25,26 Administration of proTAME resulted in the accumulation of Cyclin B1, a well-known substrate of APC (Fig. 3C). Administration of TAME also caused SENP2 accumulation and slightly decrease global SUMOylation (Fig 3C, D). However, the mRNA level of SENP2 was almost unchanged (Fig. 3E). Collectively, these data suggested that SENP2 might be a substrate of APC.
Figure 3.

SENP2 contains a canonical D-box motif. (A) Alignment of D boxes in Skp2, Cyclin B1, Securin and SENP2. (B) Alignment of D boxes in SENP2 from Mus musculus, Rattus norvegicus, Pongo abelii and Homo sapiens. (C) SHSY5Y cells were treated with 10 μM proTAME for 12 h. The whole cell lysates were immunoblotted with SENP2 and Cyclin B1 antibodies. (D) SHSY5Y cells were treated with 10 μM proTAME for 12 h. The whole cell lysates were immunoblotted with SUMO1 antibody. (E) SENP2 mRNA level from SHSY5Y cells treated with 10 μM proTAME for 12 h was detected by Realtime-PCR. The y axis represents mean ± SEM (n = 3).
SENP2 is a substrate of APCCdh1
To investigate whether SENP2 is a substrate of APC, we first tested the interaction between Cdh1 and SENP2 by coimmunoprecipitation of tagged Cdh1 and SENP2overexpressed in 293T cells. Cdh1 was able to bind to SENP2, whereas Cdc20 another coactivator of APC complex did not show any detectable association with SENP2 (Fig. 4A, Fig. S6). Endogenous Cdh1 and SENP2 could also be efficiently co-immunoprecipitated with each other in SHSY5Y cells (Fig. 4B). We further confirmed the interaction between Cdh1 and SENP2 by in vitro precipitation assays in which substantial HA-tagged Cdh1 was precipitated specifically by glutathione S-transferase (GST)-SENP2 but not by GST alone (Fig. 4C). Expression of Cdh1 but not that of Cdc20 dramatically reduced SENP2 protein level which was prevented by MG132 administration (Fig. 4D, Fig. S7). Moreover, SENP2 was degraded at a faster rate in the presence of Cdh1 (Fig. 4E). Consistent with these observations, silencing the expression of Cdh1 by siRNA or overexpression of a domination negative form of Cdh1 (DN-Cdh1) resulted in SENP2 accumulation (Fig. 4F, G). Furthermore, the half-life of SENP2 was significantly increased when Cdh1 was silenced (data not shown). Lastly, Cdh1 significantly promoted the ubiquitination of SENP2 in vivo (Fig. 4H). Taken together, our data indicated that SENP2 is a bona fide substrate of APCCdh1.
Figure 4.

SENP2 is a substrate of APCCdh1. (A) 293T cells were transfected with indicated plasmids for 36 h. Cells were lysed and subjected to immunoprecipitation with Flag M2 or HA beads. Immunoprecipitated proteins were immunoblotted with indicated antibodies. (B) Lysates of SHSY5Y cells depolarized with 50 mM KCl for 2 h subjected to immunoprecipitation with SENP2, Cdh1 or IgG antibodies. Immunoprecipitated proteins were immunoblotted with indicated antibodies. (C) Beads coated with bacterially expressed GST or GST-SENP2 were incubated with purified HA-Cdh1 protein. Beads were washed, and the bound proteins were analyzed by Western blotting with indicated antibodies. The number on the bottom of Western blot lanes indicated signal intensity of Cdh1. The signal intensity of input Cdh1 protein was set as 1. (D) SHSY5Y cells were transfected with indicated increased HA-Cdh1 plasmid for 36 h. In MG132 group, 10 μM was added 6 h before cell harvested. The whole cell lysates were immunoblotted with indicated antibodies. (E) SHSY5Y cells transfected with empty vector or HA-Cdh1 plasmids were treated with 20 μM CHX for the indicated time. The whole cell lysates were immunoblotted with SENP2 antibody. The number on the bottom of Western blot lanes indicated signal intensity of SENP2 protein against β-actin. The signal intensity of SENP2 protein against β-actin at 0 min was set as 1. (F) SHSY5Y cells were transfected with non-specific siRNA or siRNAs targeting Cdh1 for 48 h. The whole cell lysates were immunoblotted with SENP2 and Cdh1 antibodies. (G) SHSY5Y cells were transfected with empty vector or HA-DN-Cdh1 for 48 h. The whole cell lysates were immunoblotted with SENP2 and HA antibodies. (H) 293T cells were transfected with indicated plasmids for 36 h. Cells were lysed and subjected to immunoprecipitation with Flag M2 beads. Immunoprecipitated proteins were immunoblotted with indicated antibodies.
D-box-dependent degradation of SENP2 by APCCdh1
To investigate whether the degradation of SENP2 was dependent on its D-box motif, we generated mutant SENP2 protein (SENP2 Mut) in which the key arginine and leucine residues were changed into alanine and valine, respectively (Fig. 5A). SENP2 Mut was expressed at level higher than SENP2 WT and was resistant to Cdh1-mediated destabilization (Fig. 5B). Furthermore, GST–SENP2 WT but not GST–SENP2 Mut captured HA-tagged Cdh1 from SHSY5y cell extract, suggesting the D box is essential for the recognition of SENP2 by APCCdh1(Fig. 5C). To determine the importance of the D-box-mediated degradation of SENP2, we monitored the degradation rate of SENP2 WT and SENP2 Mut. In transiently transfected SHSY5y cells, the half-life of SENP2 Mut was significantly extended when compared with SENP2 WT (Fig. 5D). In agreement with these data, TAME has little effect on SENP2 Mut protein when compared with SENP2 WT protein (Fig. 5E). Cdh1 also significantly promoted the ubiquitination of SENP2WT but not SENP2 Mut in vivo (Fig. 5F). Together, these data suggested that APCCdh1 can polyubiquitinate SENP2 in a D-box-dependent manner.
Figure 5.
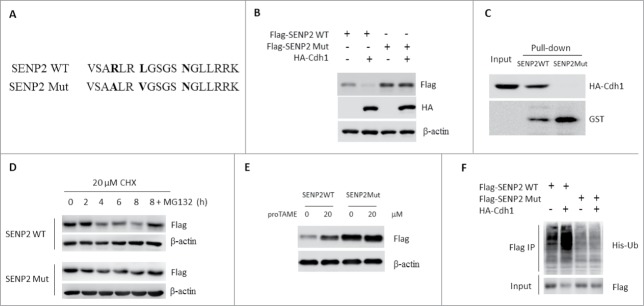
D-box-dependent degradation of SENP2 by APCCdh1. (A) Schematic diagrams of SENP2 WT and Mut. (B) SHSY5Y cells were transfected with indicated plasmids for 48 h. The whole cell lysates were immunoblotted with Flag and HA antibodies. (C) Beads coated with bacterially expressed GST-SENPWT or GST-SENP2Mut were incubated with purified HA-Cdh1 protein. Beads were washed, and the bound proteins were analyzed by western blotting with indicated antibodies. (D) SHSY5Y cells transfected with Flag-SENP2 WT or Flag-SENP2 Mut for 36 h were treated with 20 μM CHX for the indicated time. In MG132 group, 10 μM was added 6 h before cell harvested. The whole cell lysates were immunoblotted with indicated antibodies. (E) SHSY5Y cells transfected with Flag-SENP2 WT or Flag-SENP2 Mut for 36 h were treated with 10 μM TAME for 12 h. The whole cell lysates were immunoblotted with indicated antibodies. (F) 293T cells were transfected with indicated plasmids for 36 h. Cells were lysed and subjected to immunoprecipitation with Flag M2 beads. Immunoprecipitated proteins were immunoblotted with indicated antibodies.
Cdh1 regulates MEF2A transcriptional activity via SENP2 destruction
To investigate the functional significance of APCCdh1-mediated SENP2 destruction, we test whether Cdh1 regulates MEF2A transcriptional activity. Overexpression of Cdh1 but not Cdc20 dramatically repressed MEF2A transcriptional activity (Fig. 6A). Indeed, Cdh1 repressed MEF2A transcriptional activity in a dose dependent manner (Fig. 6B). Furthermore, we found that overexpression of Cdh1 also decreased KCl-induced MEF2A de-SUMOylation, decreased KCL-induced MEF2A acetylation, and KCl-induced MEF2A activation (Fig. 6C, D). Indeed, using SynGAP and Nur77 neuronal specific promoters, we found that overexpressing Cdh1 significantly decreased SynGAP- and Nur77-luciferase reporter genes activity in SHSY5Y cells treated with KCl (Fig. 6E, Fig. S8). Furthermore, silencing the expression of Cdh1 by siRNA or overexpression of DN-Cdh1 promotes the transcriptional activity of MEF2A and decreases MEF2A SUMOylation (Fig. 6F, Fig. S9). Co-expression of SENP2 Mut but not SENP2 WT significantly restored the transcriptional activity of MEF2A repressed by Cdh1 (Fig. 6G). Previous studies have showed that membrane depolarization by KCl promotes the survival of neurons, and this process is dependent on MEF2 transactivation activity.27,28 Thus, we proposed that APC/Cdh1–Senp2 axis may play a role in activity-dependent neuronal survival. To this end, we found that overexpression of Cdh1 or silencing the expression of SENP2 could prevent activity dependent neuronal survival, whereas overexpression of SENP2 or DN-Cdh1 could promote activity dependent neuronal survival (Fig. 6H, Fig. 6I), suggesting that APC/Cdh1–Senp2-MEF2 axis at least plays a role in activity dependent neuronal survival.
Figure 6.

(See previous page). Cdh1 regulates MEF2A transcriptional activity via SENP2 destruction. (A) SHSY5Y cells were transfected with MEF2A and MEF-luciferase reporter plasmids with Cdh1 or Cdc20 plasmids for 36 h. The luciferase activity was measured. Transfection efficiency was normalized by Renilla luciferase expression, and the results are presented as activation over that for empty vector transfected group. The y axis represents normalized luciferase activity ± SEM (n = 3). **represents P < 0.01 compared with Con group. (B) SHSY5Y cells were transfected with MEF2A and MEF-luciferase reporter plasmids with indicated increased Cdh1 for 36 h. The luciferase activity was measured. The y axis represents normalized luciferase activity ± SEM (n = 3). (C) SHSY5Y cells were transfected with indicated plasmids for 36 h. Cells were then treated with 50 mM KCl or 150 mM NaCl for 2 h. Cells were lysed and subjected to immunoprecipitation with Flag M2 beads. Immunoprecipitated proteins were immunoblotted with indicated antibodies. (D) SHSY5Y cells were transfected with MEF2A and MEF-luciferase reporter plasmids with indicated plasmids for 36 h. Cells were then treated with 50 mM KCl or 150 mM NaCl for 2 h. The luciferase activity was measured. The y axis represents normalized luciferase activity ± SEM (n = 3). *represents P < 0.05, **represents P < 0.01. (E) SHSY5Y cells were transfected with MEF2A and SynGap reporter plasmids with indicated plasmids for 36 h. Cells were then treated with 50 mM KCl for 2 h. The luciferase activity was measured. The y axis represents normalized luciferase activity ± SEM (n = 3). *represents P < 0.05, **represents P < 0.01. (F) SHSY5Y cells were transfected with MEF2A and MEF-luciferase reporter plasmids with siRNAs specific targeting Cdh1 for 36 h. The luciferase activity was measured. The y axis represents normalized luciferase activity ± SEM (n = 3). **represents P < 0.01 compared with Con-siRNA group. (G) SHSY5Y cells were transfected with MEF2A and MEF-luciferase reporter plasmids with indicated plasmids for 36 h. The luciferase activity was measured. The y axis represents normalized luciferase activity ± SEM (n = 3). ***represents P < 0.001 CDH1+ SENP2 Mut compared with CDH1 group. (H) Rat Primary Cerebellar Granule Neurons (CGNs) cultured in medium containing serum and depolarizing concentrations of KCl (29 mM) were transfected with indicated plasmids for 30 h. CGNs were switched to medium containing 5 mM KCl for 8 h to generate KCl withdrawal triggered CGN apoptosis model. The percentage of apoptotic cells were determined by Annexin-V/PI staining assay. The y axis represents normalized luciferase activity ± SEM (n = 3). *represents P < 0.05 compared with Con group. (I) Rat Primary Cerebellar Granule Neurons (CGNs) cultured in medium containing serum and depolarizing concentrations of KCl (29 mM) were transfected with indicated siRNAs for 30 h. CGNs were switched to medium containing 5 mM KCl for 8 h to generate KCl withdrawal triggered CGN apoptosis model. The percentage of apoptotic cells were determined by Annexin-V/PtdIns staining assay. The y axis represents normalized luciferase activity ± SEM (n = 3). *represents P < 0.05 compared with Con-siRNA group.
Discussion
By checking the protein level of SENP2 in response to activity-dependent stimuli, we have found that exogenous SENP2 was rapidly stabilized to facilitate MEF2A de-SUMOylation and activation. In this study, we mainly focused on the regulation of SENP2 by activity-dependent stimuli. SENP2 was reported to be a nucleocytoplasmic shuttling protein and ubiquitinated in the cytoplasm. We found that activity-dependent stimuli could increase the half-life of SENP2 by preventing SENP2 ubiquitination and degradation. Our earlier work revealed that activity-dependent stimuli could significantly increase global SUMOylation and alter the subcellular distribution of SUMO conjugated proteins.4 Ce´ line et al. also demonstrated that in primary cultured rat hippocampal neurons, the SUMOylation enzyme UBC9 and deSUMOylation enzymes SENP1 and SENP6 are differentially redistributed in and out of synapses upon activity-dependent stimulation.29 We then asked whether neuronal depolarization could also alter the distribution of SENP2. Indeed, SENP2 was translocated from cytosol to nucleus in response to activity-dependent stimuli, suggesting that activity-dependent stimuli prevent SENP2 ubiquitination and degradation by promoting its nuclear translocation.
We next asked which E3 ligase is required for the degradation of SENP2. Bioinformational analysis found that SENP2 contains a canonical D-box degron which is also presented in the well-known substrates of APCCdh1. This clue led us to investigate the relationship between APCCdh1 and SENP2. By using various biochemistry studies, we found that Cdh1 but not Cdc20, another APC/C activator, physically interacted with SENP2 and targeted SENP2 for ubiquitin-dependent degradation. We also test the functional significance of APCCdh1-mediated SENP2 destruction. We found that Cdh1 could enhance MEF2A SUMOylation and decrease the MEF2A transcriptional activity. Overexpression of stable SENP2 form (SENP2 Mut) could significantly restore Cdh1-repressed MEF2A activation. We also found that both Cdh1 and SENP2 were participated in activity-dependent neuronal survival. Thus, our data clarified a previous unknown Cdh1-SENP2-MEF2A axis that mediated activity-dependent stimuli-induced MEF2A transcriptional activation and activity-dependent neuronal survival.
Cdh1 is required to initiate APC activity during late mitosis and G1 and is the master of G0/G1 phase in non-neuronal cells.30,31 Recently, SENP2 was reported to play a role in the regulation of cell cycle progress.32 However, whether SENP2 itself is regulated during cell cycle progress is largely unknown. Further studies are needed to investigate the functional relevance between Cdh1 with SENP2 in the cell cycle regulation in the future.
MEF2A has been reported to be able to regulate postsynaptic dendritic morphogenesis.18 It will be interesting to test whether SENP2 or Cdh1 could play a role in postsynaptic dendritic morphogenesis. APCCdh1 complex is highly expressed in the post-mitotic neurons, which restrains the intrinsic axon growth potential.33 APCCdh1 complex is required for associative fear memory and long-term potentiation in the amygdala of adult mice.34,35 APCCdh1 complex targets both SnoN and Id2 for destruction to elevate the expression of growth inhibitory molecules results in neurite outgrowth.36,37 It will be reasonable to test whether Cdh1 plays a role in postsynaptic dendritic morphogenesis in the future. SENP2 was also reported to be highly expressed in central neuron system.13 However, the biology function of SENP2 is completely unknown in central neuron system due to the embryonic lethal of SENP2−/− mice. Indeed, several neuronal SUMOylation substrates have been identified while the de-SUMOylases specific for these substrates were remain to be identified.38 Systematic identification of the neuron specific SUMOylation substrates of SENP2 might also be required to clarify the neuronal function of SENP2. Neuron- or glial-specific knockout of SENP2 mice will be a powerful tool to investigate the neuronal function of SENP2 in the future.
Material and methods
Isolating rat neurons
All the reagents used for neurons were purchased form Invitrogen. The cortex was dissected from 10 E-18 rat embryo brains and put in a conical tube containing Hibernate®-E supplemented with 2% B-27® Serum-Free Supplement and 0.5 mM GlutaMAX™-I at 4°C. Enzymatically digest the tissue in 4 mL of Hibernate®-E medium without Ca2+ containing 2 mg/mL papain for 30 minutes at 30°C. 6 mL complete Hibernate®-E medium was added to the tube and centrifuge for 5 minutes at 150 × g. The supernatant was removed and the cells were transferred to a new tube in Neurobasal® medium with 2% B-27® Serum-Free Supplement and 0.5 mM GlutaMAX™-I for culturing.
Cell culture and treatments
Primary rat cortex neuron cells were cultured in poly-D-lysine coated 6-well plate and cultured with in Neurobasal® medium. SHSY5Y neuroblastoma cell in high passages was purchased from ATCC, and cultured in high glucose Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 2 mM glutamine and 1% antibiotics (penicillin–streptomycin). Cells were maintained at 37°C in a humidified 5% CO2 atmosphere. The cells were depolarized by KCl in the medium with addition of 31% depolarization buffer (170 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES) at a final concentration of 50 mM KCl for the indicated lengths of time. MG132 was purchased from Calbiochem, TAME hydrochloride was purchased from sigma.
Plasmids and transfection
The 3 X MRE-luc and Gal4-MEF2A expression plasmids were gifts from Dr. Michael Greenberg. The pCDNA3-MEF2A-Flag expression plasmid was a gift from Dr. Rhonda Bassel-Duby. Flag-SENP2, HA-SUMO1, His-Ubiquitin, Myc-SUMO1 and luciferase renilla reporter constructs, have been described previously. Cdh1 and Cdc20 were amplified from 293T cells by PCR and cloned into the pcDNA 3.1 vector. SENP2 MUT, SENP2 mNLS, DN-Cdh1 and DN-Cdc20 were generated using QuickChange Site-Directed Mutagenesis Kit (Stratagene). All cDNAs were completely sequenced. Cells were transiently transfected using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions.
RNAi
siRNA against SENP2 (J-006033–05) and nonspecific siRNA (D-001810–01) were purchased from Dharmacon RNA Technologies as previously described. The siRNA oligonucleotides targeting human Cdh1 (GAAGGGUCUGUUCACGUAUTT) and Cdc20 (CGGCAGGACUCCGGGCCGATT) was chemically synthesized by Dharmacon.
Nuclear-cytoplasmic fraction
The nuclear-cytoplasmic fraction of rat primary cortex neurons and SHSY5Y cells were conducted using the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific, Rockford, IL, USA) according to the manufacturer's protocol.
SUMOylation assays
In vivo SUMOylation assays were performed as described previously21. Briefly, cells cotransfected with expression plasmids for Flag-MEF2A and HA-SUMO1 were lysed in RIPA buffer (150 mM NaCl, 10 mM Na2HPO4, pH 7.2, 2 mM EDTA, 50 mM NaF, 1 mM NaVO4, 1%NP-40, 0.1% SDS, 0.75% sodium deoxycholate, 1mMPMSF, 10 mM N-ethylmaleimide). Five percent of this starting material was retained for detection of input proteins, and the remainder was subjected to immunoprecipitation overnight at 4°C. Flag M2 antibody (sigma) was used together with protein A-Sepharose beads. Immune complexes were washed 8 times with RIPA buffer at 4°C and resuspended in 2XSDS loading buffer. Immune complexes and input samples were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and probed with HA or Flag antibodies.
Immunoprecipitation (IP) assay
Immunoprecipitation experiments were performed as described previously with minor modifications21. Briefly, cells cotransfected with tagged plasmids were lysed in IP buffer (150 mM NaCl, 50 mM TrisHCl, pH 7.5, 1 mM EDTA, 50 mMNaF, 1mMNaVO4, 1%NP-40, 1mMPMSF, 10 mM N-ethylmaleimide). Five percent of this starting material was retained for detection of input proteins, and the remainder was subjected to immunoprecipitation with anti-FLAG antibodies overnight at 4°C. Immune complexes were bound to protein A-beads for 1 h at 4°C, washed twice with IP buffer, once with PBS (pH 7.4), and resuspended in 2XSDS loading buffer. Immune complexes and input samples were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and probed with appropriate antibodies
GST pull-down assay
Interaction between SENP2 and Cdh1 were assessed by GST-pull down assay. Briefly, GST or GST-SENP2 WT or GST-SENP2 MUT were expressed in BL21 cells and purified using glutathione-Sepharose beads (Amersham Pharmacia) in binding buffer (100 mM NaCl, 50 mM Tris-HCl pH 7.5, 1 mM dithiothreitol, 2 μg/ml leupeptin, 2 μg/ml aprotinin, and 100 μg/ml phenylmethylsulfonyl fluoride). After 1 h at 4°C, equal amounts of GST or GST fusion proteins were resuspended in reaction buffer (200 mM NaCl, 50 mM HEPES pH 7.5, 1 mM MgCl2, and 0.2% Triton X-100) containing 0.2 mg/ml bovine serum albumin (BSA) (Fraction V, Sigma-Aldrich) and incubated for 2 h at 4°C. Then, 1 mg HA-Cdh1-transfected cell lysate were added to each mixture followed by rotation at room temperature for 1 h, centrifugation, and 3 washes. The beads were boiled in sodium dodecyl sulfate (SDS) sample buffer to elute the bound proteins, which were resolved by 10% SDS-polyacrylamide gel electrophoresis (PAGE) followed by Western blot analysis.
Western blotting and antibodies
Protein extracts were equally loaded on 12% SDS–polyacrylamide gels and subsequently transferred to nitrocellulose membrane by electrophoresis. The membrane was blocked in Tris–buffered saline with 0.1% Tween-20 (TBST) containing 5% nonfat dried milk for 1 h at room temperature. Primary antibodies were incubated overnight at 4°C and washed 3 times each in TBST. Horseradish peroxidase-conjugated goat anti-mouse or rabbit IgG as secondary antibody was added to TBST and membrane was incubated for 1 h followed by 3 washes in TBST (5 min each). The images were visualized by FUJIFILM LAS-4000 Luminescent image analyzer. Western blot was performed using antibodies as indicated: SENP2 (H-300, Santa Cruz), SENP2 (ab3660, Abcam), SynGAP (3200s, Cell Signaling Technology), β Tubulin (ab6046, Abcam), Lamin B (C-20, Santa Cruz), Cyclin B1 Antibody (GNS1, Santa Cruz), Cdh1 (ab89535, Abcam), Cdc20 (ab26483, Abcam), Flag (M2, Sigma), Myc (ab9106, Abcam), HA (Y-11, Santa Cruz) and β-Actin (4967L, Cell Signaling Technology).
Luciferase assays
Luciferase experiments were performed as described previously with minor modifications21. Cells were transfected with firefly luciferase reporter plasmids 3XMRE-luc, MEF2A and other plasmids as indicated. For all reporter gene experiments, firefly luciferase plasmids were cotransfected with the TK-pRL vector, which expresses renilla luciferase and allows for normalization. Fresh growth media were added within 24 h of transfection. Cells were lysed 48 h after transfection. Luciferase activity was determined using the dual luciferase assay system from Promega.
Quantitative real-time PCR assays
Total RNA was isolated with use of TRIzol reagent (Invitrogen). Quantitative PCR was performed with Light Cycler 480 (Roche, Switzerland) and SYBR Green (Molecular Probes, Eugene, OR) used as a fluorescent probe. Target gene mRNA levels were normalized to that of β-actin in the same sample. The following sets of oligonucleotides were used as primers: SENP2 5′-primer (5′- GATTCCCATTCCAGCTGACCAC-3′) and 3′-primer (5′-CACTCTGATCTTTGGATAGTCA-3′) β-Actin: 5′-primer (5′- TCATGAAGTGTGACGTTGACATCC-3′) and 3′-primer (5′- CCTAGAAGCATTTGCGGTGCACGA-3′). Thermal cycling parameters were 2 min at 50°C, and 10 min at 95°C, for cDNA denaturation followed by 40 cycles of 15 sec at 95°C for denaturation, 30 sec at 60°C for anneal. For each sample, average threshold (Ct) value assays were repeated in triplicate, and the ΔCt value was determined by subtracting the average β-actin Ct value from the average SENP2 Ct value.
Immunofluorescence analysis
Cells transfected with indicated plasmids were grown on cover slips. After NaCl or KCl treatment, cells were washed with PBS, fixed with 4% formaldehyde in PBS for 10 min and permeabilized with 0.5% Triton X-100 in PBS for 10 min. Then cells were incubated with anti-Flag or HA antibodies overnight. After washing with PBS 3 times, cells were incubated with Alexa Fluor 594-conjugated goat anti-mouse IgG (1:1000; Invitrogen, Carlsbad, CA) for 1 h. DNA was stained with DAPI. Images were captured on Nikon DS-Ri1-U2 Digital Camera.
Apoptosis assay
Apoptosis was measured by the Annexin V Fluos Apoptosis detection kit (Roche Molecular Biochemicals, Mannheim, Germany) following the manufacturer's instructions. Fluorescent intensities of probes were determined by flow cytometry.
Statistical analysis
The results are presented as mean ± SEM. The Student's t test was used to compare the difference between 2 different groups. A value of P < 0.05 was considered to be statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Natural Science Foundation, Beijing, People's Republic of China, grant no. 81102513 (to Han Lu), grant nos. 81072703 and 81373492 (to Buwei Yu) and Specialized Research Fund for Outstanding Young Teachers of Higher Education, Shanghai (to Han Lu).
References
- 1. Bertrand SS, Cazalets JR. Activity-dependent synaptic plasticity and metaplasticity in spinal motor networks. Curr Pharm Des 2013; 19:4498-508; PMID:23360279; http://dx.doi.org/ 10.2174/1381612811319240014 [DOI] [PubMed] [Google Scholar]
- 2. Cerda O, Trimmer JS. Activity-dependent phosphorylation of neuronal Kv2.1 potassium channels by CDK5. J Biol Chem 2011; 286:28738-48; PMID:21712386; http://dx.doi.org/ 10.1074/jbc.M111.251942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, et al. . Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 2007; 56:517-29; PMID:17988634; http://dx.doi.org/ 10.1016/j.neuron.2007.09.032 [DOI] [PubMed] [Google Scholar]
- 4. Lu H, Liu B, You S, Xue Q, Zhang F, Cheng J, Yu B. The activity-dependent stimuli increase SUMO modification in SHSY5Y cells. Biochem Biophys Res Commun 2009; 390:872-6; PMID:19840774; http://dx.doi.org/ 10.1016/j.bbrc.2009.10.065 [DOI] [PubMed] [Google Scholar]
- 5. Carulli D, Foscarin S, Rossi F. Activity-dependent plasticity and gene expression modifications in the adult CNS. Front Mol Neurosci 2011; 4:50; PMID:22144945; http://dx.doi.org/ 10.3389/fnmol.2011.00050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Muller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin's mysterious cousin. Nat Rev Mol Cell Biol 2001; 2:202-10; PMID:11265250; http://dx.doi.org/ 10.1038/35056591 [DOI] [PubMed] [Google Scholar]
- 7. Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol 2007; 8:947-56; PMID:18000527; http://dx.doi.org/ 10.1038/nrm2293 [DOI] [PubMed] [Google Scholar]
- 8. Shin EJ, Shin HM, Nam E, Kim WS, Kim JH, Oh BH, Yun Y. DeSUMOylating isopeptidase: a second class of SUMO protease. EMBO Rep 2012; 13:339-46; PMID:22370726; http://dx.doi.org/ 10.1038/embor.2012.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Y, Dasso M. SUMOylation and deSUMOylation at a glance. J Cell Sci 2009; 122:4249-52; PMID:19923268; http://dx.doi.org/ 10.1242/jcs.050542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng J, Wang D, Wang Z, Yeh ET. SENP1 enhances androgen receptor-dependent transcription through desumoylation of histone deacetylase 1. Mol Cell Biol 2004; 24:6021-8; PMID:15199155; http://dx.doi.org/ 10.1128/MCB.24.13.6021-6028.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng J, Perkins ND, Yeh ET. Differential regulation of c-Jun-dependent transcription by SUMO-specific proteases. J Biol Chem 2005; 280:14492-8; PMID:15701643; http://dx.doi.org/ 10.1074/jbc.M412185200 [DOI] [PubMed] [Google Scholar]
- 12. Itahana Y, Yeh ET, Zhang Y. Nucleocytoplasmic shuttling modulates activity and ubiquitination-dependent turnover of SUMO-specific protease 2. Mol Cell Biol 2006; 26:4675-89; PMID:16738331; http://dx.doi.org/ 10.1128/MCB.01830-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kang X, Qi Y, Zuo Y, Wang Q, Zou Y, Schwartz RJ, Cheng J, Yeh ET. SUMO-specific protease 2 is essential for suppression of polycomb group protein-mediated gene silencing during embryonic development. Mol Cell 2010; 38:191-201; PMID:20417598; http://dx.doi.org/ 10.1016/j.molcel.2010.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chiu SY, Asai N, Costantini F, Hsu W. SUMO-specific protease 2 is essential for modulating p53-Mdm2 in development of trophoblast stem cell niches and lineages. PLoS Biol 2008; 6:e310; PMID:19090619; http://dx.doi.org/ 10.1371/journal.pbio.0060310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gong L, Yeh ET. Characterization of a family of nucleolar SUMO-specific proteases with preference for SUMO-2 or SUMO-3. J Biol Chem 2006; 281:15869-77; PMID:16608850; http://dx.doi.org/ 10.1074/jbc.M511658200 [DOI] [PubMed] [Google Scholar]
- 16. Bawa-Khalfe T, Yeh ET. SUMO losing balance: SUMO proteases disrupt SUMO homeostasis to facilitate cancer development and progression. Genes Cancer 2010; 1:748-52; PMID:21152235; http://dx.doi.org/ 10.1177/1947601910382555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rashid AJ, Cole CJ, Josselyn SA. Emerging roles for MEF2 transcription factors in memory. Genes Brain Behav 2013; 13:118-25; PMID:23790063; http://dx.doi.org/ 10.1111/gbb.12058 [DOI] [PubMed] [Google Scholar]
- 18. Shalizi A, Gaudilliere B, Yuan Z, Stegmuller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 2006; 311:1012-7; PMID:16484498; http://dx.doi.org/ 10.1126/science.1122513 [DOI] [PubMed] [Google Scholar]
- 19. Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 2006; 311:1008-12; PMID:16484497; http://dx.doi.org/ 10.1126/science.1122511 [DOI] [PubMed] [Google Scholar]
- 20. Shalizi A, Bilimoria PM, Stegmuller J, Gaudilliere B, Yang Y, Shuai K, Bonni A. PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. J Neurosci: Off J Soc Neurosci 2007; 27:10037-46; PMID:17855618; http://dx.doi.org/ 10.1523/JNEUROSCI.0361-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu H, Liu B, You S, Chen L, Dongmei Q, Gu M, Lu Y, Chen Y, Zhang F, Yu B. SENP2 regulates MEF2A de-SUMOylation in an activity dependent manner. Mol Biol Rep 2013; 40:2485-90; PMID:23224591; http://dx.doi.org/ 10.1007/s11033-012-2329-x [DOI] [PubMed] [Google Scholar]
- 22. Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol 2012; 83:1021-32; PMID:22209898; http://dx.doi.org/ 10.1016/j.bcp.2011.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barford D. Structure, function and mechanism of the anaphase promoting complex (APCC). Q Rev Biophys 2011; 44:153-90; PMID:21092369; http://dx.doi.org/ 10.1017/S0033583510000259 [DOI] [PubMed] [Google Scholar]
- 24. Almeida A. Regulation of APCC-Cdh1 and its function in neuronal survival. Mol Neurobiol 2012; 46:547-54; PMID:22836916; http://dx.doi.org/ 10.1007/s12035-012-8309-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh DC, Hathaway N, Dimova N, Cuny GD, King RW. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 2010; 18:382-95; PMID:20951947; http://dx.doi.org/ 10.1016/j.ccr.2010.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zeng X, King RW. An APCC inhibitor stabilizes cyclin B1 by prematurely terminating ubiquitination. Nat Chem Biol 2012; 8:383-92; PMID:22366722; http://dx.doi.org/ 10.1038/nchembio.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang X, She H, Mao Z. Phosphorylation of neuronal survival factor MEF2D by glycogen synthase kinase 3beta in neuronal apoptosis. J Biol Chem 2009; 284:32619-26; PMID:19801631; http://dx.doi.org/ 10.1074/jbc.M109.067785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 1999; 286:785-90; PMID:10531066; http://dx.doi.org/ 10.1126/science.286.5440.785 [DOI] [PubMed] [Google Scholar]
- 29. Loriol C, Khayachi A, Poupon G, Gwizdek C, Martin S. Activity-dependent regulation of the sumoylation machinery in rat hippocampal neurons. Biol Cell Under Auspices Eur Cell Biol Organ 2013; 105:30-45; PMID:23066795; http://dx.doi.org/ 10.1111/boc.201200016 [DOI] [PubMed] [Google Scholar]
- 30. Hu D, Qiao X, Wu G, Wan Y. The emerging role of APCCCdh1 in development. Semin Cell Dev Biol 2011; 22:579-85; PMID:21497201; http://dx.doi.org/ 10.1016/j.semcdb.2011.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manchado E, Eguren M, Malumbres M. The anaphase-promoting complexcyclosome (APCC): cell-cycle-dependent and -independent functions. Biochem Soc Trans 2010; 38:65-71; PMID:20074037; http://dx.doi.org/ 10.1042/BST0380065 [DOI] [PubMed] [Google Scholar]
- 32. Zhang XD, Goeres J, Zhang H, Yen TJ, Porter AC, Matunis MJ. SUMO-23 modification and binding regulate the association of CENP-E with kinetochores and progression through mitosis. Mol Cell 2008; 29:729-41; PMID:18374647; http://dx.doi.org/ 10.1016/j.molcel.2008.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang Y, Kim AH, Bonni A. The dynamic ubiquitin ligase duo: Cdh1-APC and Cdc20-APC regulate neuronal morphogenesis and connectivity. Curr Opin Neurobiol 2010; 20:92-9; PMID:20060286; http://dx.doi.org/ 10.1016/j.conb.2009.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pick JE, Malumbres M, Klann E. The E3 ligase APCC-Cdh1 is required for associative fear memory and long-term potentiation in the amygdala of adult mice. Learn Mem 2013; 20:11-20; PMID:23242419; http://dx.doi.org/ 10.1101/lm.027383.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pick JE, Wang L, Mayfield JE, Klann E. Neuronal expression of the ubiquitin E3 ligase APCC-Cdh1 during development is required for long-term potentiation, behavioral flexibility, and extinction. Neurobiol Learn Mem 2013; 100:25-31; PMID:23238556; http://dx.doi.org/ 10.1016/j.nlm.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wan Y, Liu X, Kirschner MW. The anaphase-promoting complex mediates TGF-beta signaling by targeting SnoN for destruction. Mol Cell 2001; 8:1027-39; PMID:11741538; http://dx.doi.org/ 10.1016/S1097-2765(01)00382-3 [DOI] [PubMed] [Google Scholar]
- 37. Lasorella A, Stegmuller J, Guardavaccaro D, Liu G, Carro MS, Rothschild G, de la Torre-Ubieta L, Pagano M, Bonni A, Iavarone A. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature 2006; 442:471-4; PMID:16810178; http://dx.doi.org/ 10.1038/nature04895 [DOI] [PubMed] [Google Scholar]
- 38. Krumova P, Weishaupt JH. Sumoylation in neurodegenerative diseases. Cell Mol Life Sci 2013; 70:2123-38; PMID:23007842; http://dx.doi.org/ 10.1007/s00018-012-1158-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.