

Within the past several years, there have been major advances in cancer treatment through the use of molecularly targeted therapy, though resistance to therapy remains a significant issue. The clinical success of driver oncogene targeted therapy is limited by both innate and acquired resistance that occurs in most patients.1,2 Understanding the molecular basis of resistance is a cardinal goal of modern molecular oncology because the knowledge gained offers insight into cancer cell signaling and therapeutic strategies to suppress resistance and improve patient survival.
We recently investigated the biological basis of resistance to targeted therapies against aberrant RAS-RAF-MEK-ERK (MAPK, mitogen activated protein kinase) signaling in human cancer,3,4 as RAF-MEK targeted therapy resistance is a major clinical problem. RAF-MEK inhibitor treatment is initially (but only temporarily) effective in some but not all BRAF-mutant patients, and is largely ineffective in RAS-mutant patients.5 Through a genetic screen in human BRAFV600E mutant NSCLC cells, we defined genes that when suppressed enhanced response to the BRAF inhibitor vemurafenib, the first mutant BRAF targeted therapy clinically approved.5 Our studies showed that the Hippo pathway effector YAP1 (yes-associated protein 1) functioned as a parallel survival input to promote resistance to RAF-MEK inhibitor therapy (Fig. 1).3 Co-inhibition of YAP and RAF or MEK was synthetically lethal in several BRAF mutant tumor types (melanoma, lung, colon, thyroid) and also in RAS mutant tumors (lung, melanoma, pancreas). YAP mediated resistance by regulating the levels of the anti-apoptotic protein BCL-xL (BCL2L1) together with MAPK signaling. BCL-xL levels were suppressed to a degree sufficient to trigger apoptosis only upon combined YAP and RAF-MEK inhibition, establishing the importance of this dual regulation of BCL-xL. Additionally, we found that increased YAP protein expression in BRAFV600E patient tumors was a biomarker of worse initial response and also of acquired resistance to RAF and MEK inhibition in patients, establishing the clinical relevance of our findings. The findings uncover the synthetic lethality of YAP and RAF-MEK co-inhibition as a potentially effective approach to improve response and patient survival.
Figure 1.
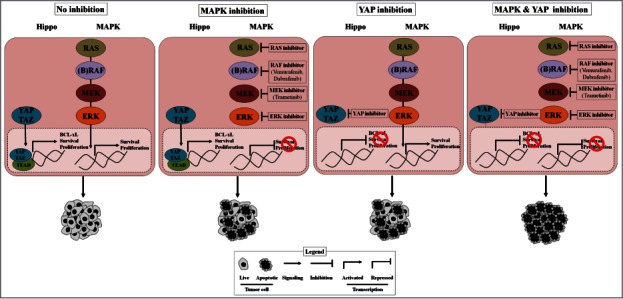
YAP promotes cancer cell survival and resistance to RAF-MEK-targeting agents. Shown is schematic of the role of YAP in BRAF- and RAS-mutant tumor cells and the potential integrative function of YAP in resistance to MAPK pathway inhibitors. YAP inhibition along with MAPK pathway inhibition enables complete anti-tumor response, in contrast to either YAP inhibition or MAPK pathway inhibition alone. MAPK, mitogen activated protein kinase; YAP, yes-associated protein.
The findings in our study prompt additional areas for future investigation. The data raise the possibility that YAP may enable survival and escape from other targeted therapies against oncogenic drivers that promote MAPK pathway activation. For example, YAP might regulate response to inhibition of mutant receptor tyrosine kinases (RTKs) such as the epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) that drive the growth of many lung adenocarcinomas and that are targeted with approved EGFR and ALK kinase inhibitors, respectively.1
The findings further suggest that investigation of the molecular basis of the crosstalk between YAP and MAPK pathway signaling is warranted. Our data indicate that many signaling components, including BCL-xL, are co-regulated by these pathways in BRAF mutant cancer cells. The underlying molecular events that control this co-regulation and the extent to which tissue- or genetic driver-specific regulatory processes modulate this crosstalk are unclear and avenues for future studies.
As YAP is a potential target across many tumor types,6 pharmacologically blocking YAP is an important and ongoing challenge. YAP inhibitors have been reported,6 but whether these agents suppress the pathway potently and selectively to enable a sufficient therapeutic window in patients remains unclear. The development of more specific drugs that inhibit YAP may be necessary for clinical translation to test the emerging hypothesis that YAP blockade may have therapeutic efficacy in selected cancer patients, including (but not limited to) those with BRAF and RAS mutant cancers.
Several mechanisms of resistance to RAF and/or MEK inhibition have been identified.7 In melanoma, most mechanisms uncovered to date have been implicated primarily in the setting of acquired resistance.7 These resistance mechanisms fall into two broad classes: (1) alterations that re-activate MAPK pathway components during RAF inhibitor therapy, such as alternatively spliced forms of mutant BRAF, BRAF amplification, and activating mutations in MEK or NRAS; and (2) upstream RTK activation via receptor or ligand upregulation (for example, EGFR) that bypasss the effects of RAF (or MEK) inhibitor therapy by activating multiple downstream survival pathways. Our findings have implications for combating each of these molecular roads to resistance, as YAP may play a broad contributory role to resistance across both molecular classes (Fig. 1). Additional studies in patient-derived cell lines with these other resistance mechanisms present would help elucidate the role of YAP in each.
As YAP can limit response even with maximal MAPK pathway suppression, combined YAP and RAF-MEK-ERK inhibition may be necessary to achieve definitive therapeutic efficacy. Further, many YAP target genes include receptors and their ligands (for example, epidermal growth factor).3 YAP might therefore additionally contribute to RTK bypass signaling. Hence, we propose synthetic lethal upfront co-inhibition of YAP and MAPK pathway signaling is an attractive potential solution to more definitively combat resistance, particularly since upfront co-inhibition of RAF plus MEK or RAF plus RTK (EGFR) inhibitor treatment remains plagued by acquired clinical resistance. Integrative basic and translational studies will further define the role of YAP in cancer biology and therapy, with important implications for improving cancer patient outcomes.
References
- 1. Rosell R, et al. Lancet 2013; 382:720-31; PMID:23972815; http://dx.doi.org/ 10.1016/S0140-6736(13)61715-8 [DOI] [PubMed] [Google Scholar]
- 2. Sawyers CL, van 't Veer LJ. Clin Cancer Res: Off J Am Assoc Canc Res 2014; 20:4978-81; PMID:25204554; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-2295 [DOI] [PubMed] [Google Scholar]
- 3. Lin L, et al. Nat Genet. 2015 Mar; 47(3):250-6; PMID:25665005; http://dx.doi.org/ 10.1038/ng.3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin L, et al. Proc Natl Acad Sci U S A 2014; 111:E748-757; PMID:24550319; http://dx.doi.org/ 10.1073/pnas.1320956111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bollag G,, et al. Nature 2010; 467:596-9; PMID:20823850; http://dx.doi.org/ 10.1038/nature09454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moroishi T, et al. Nat Rev Cancer 15:73-9; PMID:25592648; http://dx.doi.org/ 10.1038/nrc3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sullivan RJ, Flaherty KT. Brit J Dermatol 2014; 170:36-44; PMID:24443912; http://dx.doi.org/ 10.1111/bjd.12698 [DOI] [PubMed] [Google Scholar]