

Abstract
Spatially distinct pools of the small GTPase Cdc42 were observed, but the major focus of research so far has been to investigate its signaling at the plasma membrane. We recently showed that the Golgi pool of Cdc42 is relevant for cell polarity and that it is regulated by GM130, a Golgi matrix protein. Loss of GM130 abrogated cell polarity and consistent with the notion that polarity is frequently impaired in cancer, we found that GM130 is downregulated in colorectal cancer. Whether the loss of GM130 solely affects polarity, or whether it affects other processes relevant for tumorigenesis remains unclear. In a panel of breast cancer cells lines, we investigated the consequences of GM130 depletion on traits of relevance for tumor progression, such as survival, proliferation, adhesion, migration and invasion. We show that cellular assays that depend on polarity, such as chemotaxis and wound scratch assays, are only of limited use to investigate the role of polarity modulators in cancer. Depletion of GM130 increases cellular velocity and increases the invasiveness of breast cancer cells, therefore supporting the view that alterations of polarity contribute to tumor progression.
Introduction
The Ras superfamily of small GTPases is composed of five families and Rho GTPases are one of these families that comprises at least 22 members.1 Except few members, the major part of Rho family GTPases acts as molecular switches that cycle between the active (GTP-bound) and inactive (GDP-bound) state.1,2 Activation of Rho GTPases is mediated by one of about 60 guanine nucleotide exchange factors (GEFs), which exchange GDP for GTP. Deactivation is mediated by one of about 70 GTPase activating proteins (GAPs), which stimulate hydrolysis of GTP to GDP. Rho GTPases are expressed in all eukaryotes and they function as key regulators of the cytoskeleton and membrane traffic, thereby modulating cell migration and polarization. The most studied Rho family GTPases are RhoA, Rac1 and Cdc42, which have been almost exclusively studied in the context of signaling at the plasma membrane. With respect to cell polarity, Cdc42 appears to take a center stage,3 but again our understanding of its role in cell polarity is based on research focusing on Cdc42 signaling at the plasma membrane. For instance, during chemotaxis phosphoinositide 3-kinase activates Cdc42 at the leading edge.4 Receptor tyrosine kinases recruit GEFs for Cdc42 and activate it at the cell surface.5 Active Cdc42 at the leading edge will then signal via the Par complex to activate GSK-3β and will result in stabilization of microtubule plus ends at this plasma membrane subdomain.6 However, the plasma membrane is not the sole location of Cdc42, which has been detected on endomembrane locations and most prominently at the Golgi apparatus.7,8 The functional significance of this spatial pool of Cdc42 at the Golgi remained unclear. We recently used fluorescence resonance energy transfer (FRET) microscopy to show that Cdc42 is active at the Golgi and that this pool is important for cell polarization.9 The Golgi apparatus is increasingly viewed as a platform for the spatial regulation of signaling molecules10,11 and its role in cell migration and related processes such as metastasis is becoming increasingly evident.12 We showed that the Golgi-matrix protein GM130, regulates Cdc42 specifically at the Golgi without affecting plasma membrane Cdc42. The effect of GM130 towards Cdc42 was dependent on RasGRF, which we identified as a new interaction partner for GM130 (see schematic in Fig. 1A). The GM130-RasGRF interaction was not only important for the regulation of Cdc42, but it also controlled the level of active Ras, thereby providing an additional example for crosstalk of small GTPases.13 Since the balance between Ras and Cdc42 signaling is important to maintain epithelial morphogenesis, we reasoned that GM130 might be lost in human tumors. Indeed, GM130 was progressively lost when comparing healthy colon with adenoma and adenocarcinoma of the large intestine.9 Thus, we proposed that spatial regulation of Cdc42 by GM130 is relevant for cell polarity, and thereby to cancer progression. This is based on the notion that defects in cell polarity act as catalyzers of tumorigenesis and metastasis. However, it is not clear what cancer-relevant cellular traits are induced by GM130 depletion. Here, we further investigated the role of GM130 in cancer with a focus on breast cancer. We explored a panel of breast cancer cells comparing the levels of GM130 and their correlation with Golgi morphology. Furthermore, we tested the effect of GM130 depletion on cancer-relevant traits such as proliferation and apoptosis. Finally, we determined the effect of GM130 depletion on cell migration and found that loss of this Golgi-matrix protein inhibits directed motility, while at the same time increasing random cell motility. These results further support the notion of an important role of GM130 in cancer and point to the fact that loss of polarity genes might be of greater relevance for cancer. Our results also indicate that the use of assays that are dependent on cell polarization (e.g., wound scratch assay) might not be useful to predict the tumorigenic potential of alterations of proteins involved in cell polarity.
Figure 1.
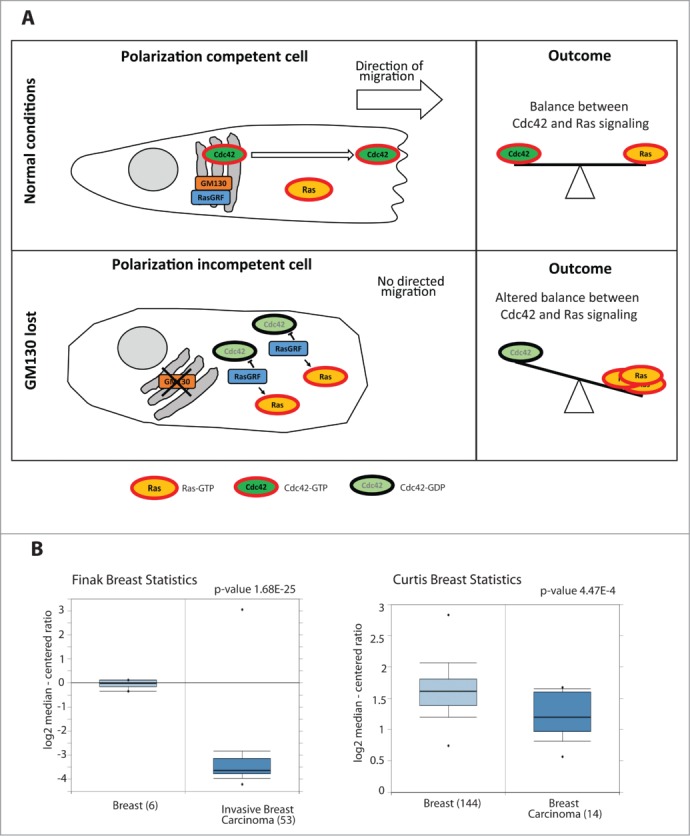
(A) Schematic representing the mechanism of action of GM130: GM130 binds to RasGRF and blocks its function. Once Cdc42 is activated, it accumulates on membranes and the Golgi sends Cdc42 in a polarized fashion to the Leading Edge of the migrating cell, thereby conferring persistence to the migration. GM130 will therefore contribute to maintain the balance between Cdc42 and Ras signaling. When GM130 is lost, the cell cannot migrate persistently and there is an imbalance between Cdc42 and Ras signaling. (B) Box Plots of two studies 32,33 comparing the mRNA levels of GM130 in normal tissues and in breast cancer tissues (obtained from Oncomine).
Results and Discussion
GM130 expression in breast cancer
We showed previously that GM130 expression is reduced in colorectal cancer.9 Searching the Oncomine database (https://www.oncomine.org), we found several studies that reported lower GM130 mRNA levels in breast cancer compared to normal mammary tissue and two examples are displayed in Figure 1B. Thus, the defect in polarity imposed by GM130 depletion potentially represents a selective advantage to breast cancer, which is in line with findings of others who showed that loss of polarity genes promotes tumor progression.14-16 We compared the levels of GM130 mRNA and protein expression in a panel of breast cancer cells (five basal and four luminal cell lines). There was no significant difference between the two subtypes with respect to GM130 levels (Fig. 2A and B). Interestingly, while most cell lines had relatively comparable amounts of GM130 mRNA (Fig. 2B), the variability in the levels of GM130 protein was more pronounced (Fig. 2A) and did not correlate with the differences observed at the mRNA level. For instance while BT20 cells had higher GM130 mRNA levels than BT549 cells, the situation was opposite at the protein level. This is important for the future when comparing studies that rely on measurements of mRNA (qPCR or microarray) vs. those that rely on protein measurements (immunoblots or immunohistochemistry). Two possible explanations are either that the GM130 protein is subject to different rates of degradation or that these cells have different rates of translating the GM130 mRNA. Elucidating whether these scenarios apply will be important in the future to better understand the regulation of GM130 in tumors.
Figure 2.

(A) Histogram representing the quantification of GM130 levels compared to actin, obtained from 3 independent western blots. Results are showed as averages ± SE. Below, a representative western blot. (B) mRNA levels of GM130 normalized to GAPDH mRNA. Results are shown as 1/(CtGM130-CtGAPDH). Bar graphs are averages of 3 independent experiments ± SE. GAPDH was amplified at cycle 15.8687 ± 0.3265, confirming that it can be considered an housekeeping gene also when comparing different cell lines. (C) The Golgi Compactness Index (GCI) was calculated as described in the materials and methods. More than 20 cells per experiment in three independent experiment were scored for each cell line. Results are shown as averages ± SE. (D) GCI was plotted on the x axis, the average of the protein levels of GM130 was plotted on the y axis. The linear correlation between these two parameters was assessed for all data points (black), only for luminal cell lines (blue) or only for basal cell lines (red).
Golgi morphology in breast cancer cells
Since cancer cells were shown to often display altered Golgi structure,17 we determined the Golgi compactness index as described previously.18 Some cell lines displayed strongly fragmented Golgi phenotypes, while others exhibited the Golgi as a single copy organelle located in the center of the cell (Figs. 2C and 3). Overall, there was a weak correlation (R2 = 0.42) between the expression levels of GM130 and the degree of Golgi compactness (Fig. 2D). However, when the two subtypes were analyzed separately we observed that luminal cell lines have a stronger correlation between Golgi compactness and GM130 levels, while basal cell lines displayed a very weak correlation. In the future, an analysis of a considerably larger collection of breast cancer cells must be performed to correlate GM130 levels and Golgi morphology.
Figure 3.
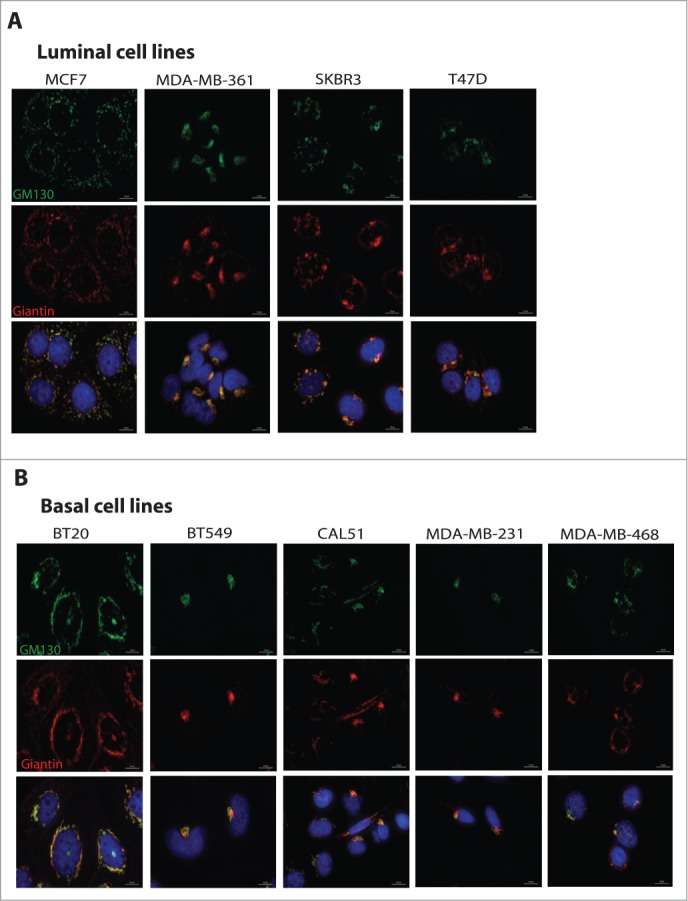
Cells were plated on coverslips and processed for immunofluorescence staining using antibodies against GM130 (green) and Giantin (red) to visualize the Golgi, and DAPI to stain the nuclei. Scale bars, 10 μm. (A) Representative images of Golgi in luminal cell lines. (B) Representative images of Golgi in basal cell lines.
GM130 depletion does not affect proliferation, apoptosis or adhesion
Based on our previous work,9 we speculate that the effect of GM130 loss is that it de-regulates cell polarity and thereby promotes tumorigenesis. However, we did not consider the possibility of effects on other cancer hallmarks such as proliferation, apoptosis or adhesion.19 Such effects could occur either as a consequence of the loss of polarity, or might alternatively emerge downstream of alterations distinct from the defect of polarity,20,21 but in any case it is important to determine the effect of GM130 knockdown on these processes. We therefore stably depleted GM130 in two luminal (MCF7 and T47D) and two basal (MDA-MB231 and BT549) cell lines (Fig. 4A). In none of the cells could we detect an appreciable effect on adhesion or proliferation (Fig. 4B and C). In the case of apoptosis, we compared the basal level of apoptosis as well as cell death induced by the cytostatic drug Doxorubicin. In all cell lines, we observed a significant increase in apoptosis after treatment with the cytostatic agent (Fig. 4D). We next compared the sensitivity towards Doxorubicin treatment in mock-depleted and GM130-depleted conditions. In T47D cells, GM130 knockdown led to a reduced sensitivity towards Doxorubicin treatment, although the result was statistically significant, it was very weak. In MDA-MB231 cells, depletion of GM130 increased sensitivity and this effect was not only statistically significant, but was also strong. One possible explanation is that MDA-MB231 cells have low GM130 protein levels compared to the other three cells lines tested (Fig. 2A) and therefore, we might speculate that these cells are more sensitive to depletion of this Golgi matrix protein. Another possibility is that MDA-MB231 are transformed due to a mutation in K-Ras, while the other cell lines are transformed due to mutations in the PI3-kinase pathway (see Table 1). However, this remains only speculative at the current state and requires future investigations.
Figure 4.
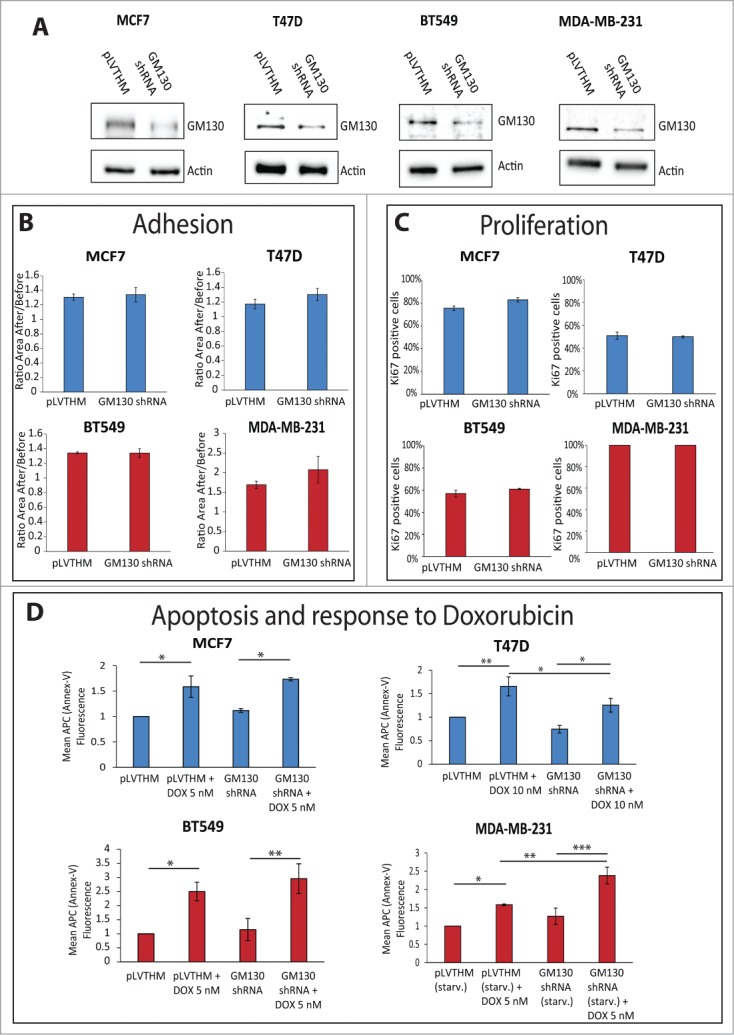
(See previous page). (A) The indicated cell lines were stably transduced with either a control plasmid (pLVTHM) or a plasmid encoding a shRNA against GM130 (GM130 shRNA). The cells were lysed and processed for western blot with antibodies against GM130 and actin to verify the efficiency of the shRNA against GM130. (B) The capacity of the indicated cells to adhere to a soft substrate (Collagen type IV) were assessed as described in the materials and methods. Results are shown as averages of three independent experiments ± SE. No significant difference was observed using the Student T test. (C) Cells were plated on coverslips, grown to subconfluency and processed for immunostaining against the proliferative marker Ki67 and DAPI. The percentage of cells positive for Ki67 was calculated counting at least 300 cells per experiment in three independent experiments. Results are shown as averages ± SE. No significant difference was observed using the student T test. (D) 200.000 cells were plated on 6-well plates. The next day, cells were either left untreated or treated with the indicated concentration of doxorubicin overnight. MDA-MB231 were also left overnight in the absence of FCS, in addition to the addition of doxorubicin. Cells were then processed for Annexin-V-APC as described in materials and methods. The mean fluorescence of the Annexin-V staining was measured in at least three independent experiments for every condition. Results are shown as averages ± SE. Asterisks indicate statistically significant differences calculated with ANOVA using the Newman-Keuls correction for multiple comparisons (*P < 0.05; **P < 0.01, ***P < 0.001).
Table 1.
Main characteristics of the cell lines analyzed
| Cell line | Type | ER | PR | ERBB2 | Mutations | Ref. |
|---|---|---|---|---|---|---|
| MCF7 | Luminal | + | + | − | PI3KCA | 25,27-29 |
| MDA-MB-361 | Luminal | + | + | − | PI3KCA | 25,27,29 |
| SKBR3 | Luminal | − | − | + | TP53 | 25,27,30 |
| T47D | Luminal | + | + | + | PI3KCA | 25,27,29 |
| BT20 | Basal | − | − | − | CDKN2A, PI3KCA, TP53 | 25,27 |
| BT549 | Basal | − | − | − | PTEN, RB1, TP53 | 25,27,31 |
| CAL51 | Basal | − | n.a. | + | PI3KCA | 28,31 |
| MDA-MB-231 | Basal | − | − | − | BRAF, CDKN2A, KRAS, NF2, TP53, PDGFRA | 25,27,31 |
| MDA-MB-468 | Basal | − | − | − | PTEN, RB1, SMAD4, TP53 | 25,27,31 |
Effect of GM130 depletion on cell motility
We finally tested the effects of GM130 depletion on cell migration. A classic is to perform wound-scratch assays and as shown previously for HeLa cells,9 directed motility of four breast cancer cell lines was inhibited by GM130 depletion (Fig. 5A). This is consistent with the defect in cell polarization imposed by GM130 depletion. Furthermore, we performed an invasion assay wherein cells had to invade through extracellular matrix (Geltrex) towards a gradient (serum). In agreement with a polarization defect, GM130 knockdown reduced the invasive capacity of cells (Fig. 5B). However, loss of polarity genes was previously shown to promote metastasis,14,15 and GM130, which we identified as a polarity regulator, also appears to be downregulated in breast (Fig. 1B) and colonic9 cancer. It is difficult to reconcile a pro-tumorigenic effect with an inhibition of invasion. However, we stress that the wound scratch assay and invasion assays towards a chemoattractant are strictly dependent on polarity and therefore are of limited use to evaluate the role of polarity proteins in cancer. Using intra-vital microscopy, it was shown recently that tumors exhibit two populations of cells, one that is fast moving and one that is slow.22 There, it was hypothesizes that the fast moving cells are those who cover large distances until they reach the proximity of a vessel, where they become slower. Another report showed that cancer cells that move faster are those which are more likely to metastasize, as they give rise to more circulating tumor cells.23 We therefore tested the effect of GM130 depletion on random motility of two breast cancer cells, BT549 and MDA-MB231, both of which are of the basal subtype, which is known to give rise to more metastasis than luminal cells. In BT549 cells, we observed a robust increase in a random motility assay (Fig. 5C). The magnitude of the effect of GM130 depletion is in the range of what has been observed for other genes of relevance to cell motility.24 Of note, persistency of cell movement was reduced by GM130 knockdown (Fig. 5C). A similar observation has been made for HeLa cells migrating on collagen (not shown). In MDA-MB231 cells, velocity tended to increase by GM130 depletion, but the effect did not reach statistical significance and was weaker compared to BT549 cells (Fig. 5C). This is potentially due to the fact that MDA-MB231 cells express a constitutively active K-Ras oncogene (Table 1). Hyperactive Ras signaling will over-activate ERK1/2, which is a positive driver of cell movement. In addition, GM130 depletion itself hyperactivates ERK1/2 9. Therefore, it might not be possible to detect the contribution of GM130 towards motility in cells with an already highly active Ras-ERK pathway. Finally, we wanted to determine the effect of GM130 depletion on invasion using an experimental setting that does not rely on a gradient. Therefore cells were allowed to invade into matrix, but the upper and lower changer in this trans-well assay were identical with respect to the amount of serum (see schematic in Fig. 5D). Strikingly, GM130 depletion strongly increased the invasive capacity of BT549 cells, but did not exert the same effect on MDA-MD231 cells (Fig. 5D). We think that the increased invasion of BT549 cells is linked to the increase in cell motility (Fig. 5C). Consequently, MDA-MB231 cells, which do not migrate faster after GM130 depletion, also do not invade better. We speculate that this could be due to the fact that MDA-MB231 cells are KRas-transformed. In addition, these cells have relatively low levels of GM130 compared to BT549 cells, and therefore, might be more sensitive to depletion of this Golgi-matrix protein. This latter assumption is supported by the observation that MDA-MB231 cells are the only one that are sensitized to undergo apoptosis upon GM130 knockdown (Fig. 4D). Luminal cells are less invasive than basal cells. Nevertheless, T47D were shown to be slightly invasive25 and when we tested the effect of GM130 depletion in this cell line and found that the number invading cells doubled compared to control (data not shown). This result is in line with the observation made in BT549 cells that silencing GM130 results in increased invasiveness.
Figure 5.
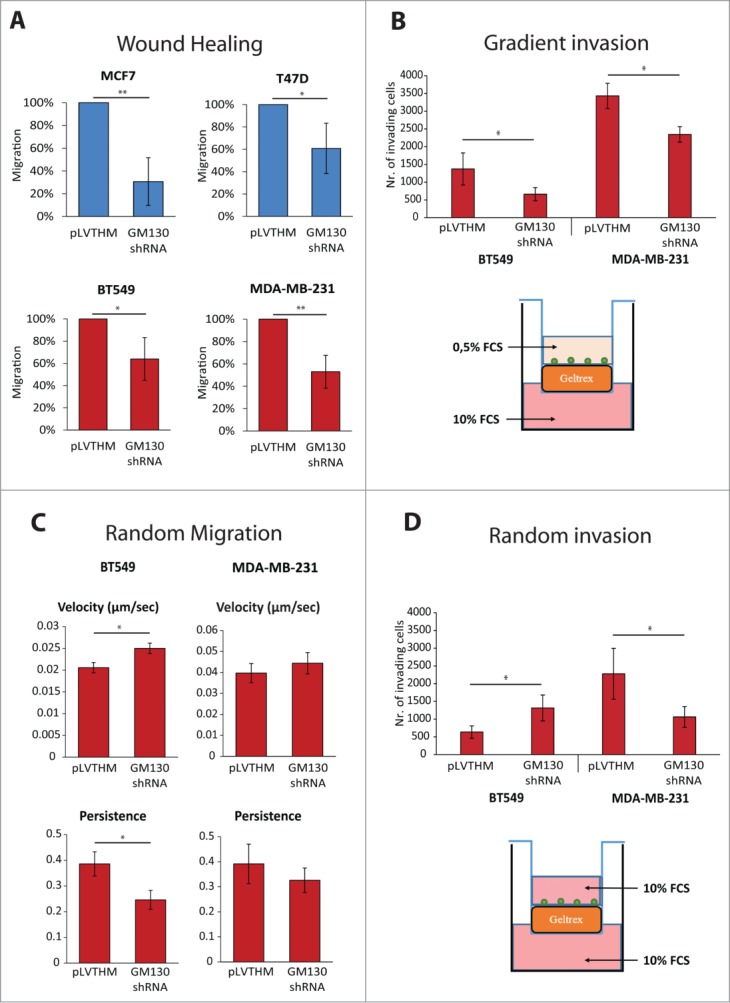
(See previous page). (A) Wound healing assays were performed as described in materials and methods. The area migrated by the cells was measured. Results are expressed as percentage of migration compared to control conditions. Results are shown as averages of at least three independent experiments ± SE. Asterisks indicate statistically significant differences calculated using Student T test (*P < 0.05; **P < 0.01). (B) Cells were plated on top of Geltrex covered membranes with 8 μm pores and let invade in the presence of a chemotactic gradient (FCS) for 24 h. The number of cells which invaded through Geltrex was then counted. Results are shown as averages of at least three independent experiments ± SE. Asterisks indicate statistically significant differences calculated using Student T test (*P < 0.05). Below the bar-graph, schematic of the experimental settings. (C) Cells were plated on glass bottom slides and imaged overnight. Velocity and persistence were calculated as described in the materials and methods. Results are shown as averages of three independent experiments ± SE. Asterisks indicate statistically significant differences calculated using Student T test (*P < 0.05). (D) Cells were plated on top of Geltrex covered membranes with 8 μm pores and let invade in the absence of a chemotactic gradient for 24 h. The number of cells which invaded through Geltrex was then counted. Results are shown as averages of at least three independent experiments ± SE. Asterisks indicate statistically significant differences calculated using Student T test (*P < 0.05). Below the bar-graph, schematic of the experimental settings.
Our results indicate that GM130, a protein we identified to regulate cell polarity,9 is frequently lost in breast and colon cancer. This further strengthens the notion of the relevance of polarity regulators in cancer. In addition, our results raise concerns as to the choice of assay for how to evaluate the relevance of a polarity gene in cancer. The widely used wound scratch assay suffers from being dependent on cell polarity and therefore, defects in cell polarity will inhibit migration in this assay. However, this result will not necessarily mirror the role of the examined proteins in vivo, where they might even enhance migration. In addition, the classical trans-well invasion assays are typically performed using a chemotactic gradient and are thus also polarity dependent. This assay probably does not measure invasion per se, but rather chemotactic invasion. We propose that it will be important in the future to perform invasion assays without gradients. The marked difference in the invasive capacity of GM130 depleted cells in the gradient-driven (reduced invasion) vs. the non-gradient assay supports this claim.
Materials and Methods
Cell Culture and transfection
HeLa, MCF7, MDA-MB-361, T47D cells were cultured in Dulbecco's modified Eagles medium (DMEM) supplemented with 10% FCS and 100 U/ml penicillin/streptomycin. MDA-MB-468, SKBR3, CAL51 were cultured in Dulbecco's modified Eagles medium (DMEM) supplemented with 20% FCS and 100 U/ml penicillin/streptomycin. BT20 were cultured in Advanced MEM supplemented with 10% FCS, 2mM ultraglutamine, and 100 U/ml penicillin/streptomycin. MDA-MB-231 and BT549 were cultured in RPMI supplemented with 10% FCS and 100 U/ml penicillin/streptomycin. To generate stable cell lines, cells were transduced with lentiviruses (produced according to the protocols of Trono's lab) and transduced cells were either selected with antibiotics or sorted via FACS (using a BD FACSAriaTM cell sorter).
Plasmids
Lentiviral packaging plasmids were bought from Addgene (psPAX2 nr. 12260, pMD2.G nr. 12259 and pLVTHM nr. 12247, from Didier Trono's laboratory). GM130 GIPZ shRNA was bought from Thermo Scientific (Clone ID: V3LHS_313361).
Antibodies and immunofluorescent labels
Mouse monoclonal anti-GM130 antibody was from BD-Biosciences (WB dilution: 1:250 in PBS-BSA 5% overnight – IF dilution 1:1000 in PBA-BSA 3% for 1h at RT – Cat nr. 610823). Rabbit polyclonal anti-Giantin antibody was from Covance (IF dilution 1:1000 in PBS-BSA 3% for 1h at RT – Cat nr PRB-114C). Rabbit polyclonal anti-Ki67 antibody was from Abcam (IF dilution 1:1000 in PBS-BSA 3% – Cat nr. 15580). For immunofluorescence staining cells were grown on glass coverslips and then fixed with 3% paraformaldehyde for 10 min at RT followed by permeabilization with PBS containing 3% BSA and 0.2% triton X100. DAPI Fluoromount G (Southern biotech – Cat nr. 0100-20) was used to mount the coverslips.
Confocal microscopy and image analysis
Live Microscopy was performed on a LeicaSP5 confocal laser scanning microscope using a 10X objective (NA 0.3) equipped with a Ludin Chamber. Alternatively, a Zeiss Axiovert 200M with 37°C incubation chamber was used. A 10X objective (NA 0.45) was used for live images with the Zeiss system. Fluorescent Images were acquired on the Zeiss Axiovert 200M using 20X objective (NA 0.4) or a 100X oil immersion objective (NA 1.4). ImageJ was used to process the images. Briefly, the process “subtract background” was used on every image. If needed to improve visibility, brightness and contrast of the images were corrected (linearly) using the “adjust” menu of ImageJ. Images coming from the same experiment were all modified the same way to leave unaltered the information. Merged images were created with the appropriate function of ImageJ, after correcting the single channel images as described above. The Golgi compactness index was calculated using the formula GCI = 4π × Area/Σ Perimeter2 as described in Bard et al.,18 with GM130 as Golgi marker.
Wound Assays
Cells were grown on glass coverslips, or on 8 wells glass bottom chambers (BD Falcon Cat Nr. 354108), or on 4 well glass bottom chambers (Lab Tek Cat Nr. 177399). Once confluency was reached, a wound was made using a 10 μm pipette tip. HEPES at a final concentration of 100mM was added to the cells before starting the overnight imaging. One image every 5 minutes was taken using transmitted light for at least 8 hours. Alternatively, wound scratches were imaged immediately after scratching and after overnight migration. The area covered by the cell sheets during migration was measured. The area migrated by control cells was set as 100% and the other values were calculated subsequently. At least three matched experiments (control and GM130 shRNA cells) per conditions were performed.
Random Migration
2E4 cells were plated per well in an 8-well glass bottom chamber slide (BD Falcon Cat Nr. 354108) or alternatively on glass bottom dishes (MatTek corp. P35G-1.5-20-C). Imaging was started after an appropriate time to allow cells to adhere (at least 4 h after plating). HEPES at a final concentration of 100mM was added to the cells before starting the imaging. 1 image every 5 minutes was acquired using transmitted light for at least 8 hours. Individual cells were manually tracked with the ImageJ plugin MTrackJ. Average velocity is automatically given by MTrackJ. Persistency was calculated using the formula: D2S/LEN. D2S = linear distance between first and last position, LEN = total length of the track. At least 5 cells/experiment were tracked in every condition, in at least 3 different experiments.
RT-PCR
Total RNA was isolated from cells by using the Qiagen RNeasy kit. cDNA was prepared from 500 ng total RNA by using the high capacity cDNA reverse transcription kit (Applied Biosystems) following manufacturers’ instructions. Q-PCR was performed by using Fast SYBR green PCR MasterMix (Applied Biosystems). RT-PCR were run on 7900HT Fast RT-PCR system (Applied Biosystems). Expression of GM130 was normalized to the expression of GAPDH. Specific primers for GM130 were designed with the online software Primer3Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). Specific primers for GAPDH were previously described 26. Sequences are:
GAPDH: F: 5′ CGCTCTCTGCTCCTCCTGTT 3′ - R: 5′ CCATGGTGTCTGAGCGATGT 3′ - GM130: F: 5′ TGCCAGTCTCACTAGCATGG 3′ - R: 5′ TGACACATGTCGCAGACTCA 3′.
Adhesion assay
The adhesion assay was performed as described previously.15 Briefly, glass coverslips or glass bottom dishes were coated with tail vein rat collagen type IV at a concentration of 2 mg/ml (Roche Cat nr. 11 179 179 001). Surfaces were covered with collagen and air dried followed by washing with PBS and storage at +4°C. Cells were trypsinized and counted. Imaging was performed on a LeicaSP5 confocal laser scanning microscope using a 10X objective (NA 0,3) at 37°C. Imaging was started immediately after plating cells onto the glass bottom dish. The time point when cells settled on the bottom of the coverslip was used as time point 0. One image was acquired every 10 minutes until cells started to adhere on the substrate. The final time point was chosen according on when the morphological changes of control transduced cells became evident. This corresponded to 80 minutes after plating for MCF7 and BT549, 50 minutes for MDA-MB-231 and 460 minutes for T47D. The total area occupied by the cells present in one field of view was measured with ImageJ at the time point 0 and at the final time point. The ratio between the area at the final time point and the area at time point 0 was then calculated (Area After/Before). At least 3 matched experiments (control and GM130 shRNA cells) per condition were performed.
Apoptosis assay
3E5 cells were plated on 6 well plates. 24 h later, Doxorubicin was added to the cells at the minimum concentration required to induce apoptosis (determined experimentally). Cells were stained with Annexin V-APC (BD Pharmingen – Cat nr. 550474) according to manufacturer's instructions. 24 h later, cells were trypsinized, resuspended in 1ml of medium, washed with ice cold PBS, resuspended in binding buffer (0.1 M Hepes (pH 7.4) 1.4 M NaCl, 25 mM CaCl2) at a density of 1E6 cells/ml. 1E5 cells were then mixed with 5 μl of Annexin-V-APC (BD Pharmingen – Cat nr. 550474) and incubated at RT for 15 minutes. Then, flow cytometry analysis was performed on a BD LSR II. At least 3 matched experiments per conditions were performed.
Proliferation assay
1.5E5 cells were plated on glass coverslips. 48 h later, cells were fixed with PFA 3% for 10 minutes and subsequently stained with an anti Ki67 antibody and DAPI. Images were acquired with the Zeiss Axiovert 200 m using a 20x objective. 3 to 4 images per condition were taken. Cells positive for Ki67 were counted manually in ImageJ with the “cell counter” plugin and compared with the total number of cells (counting of DAPI stainings). At least 300 cells per experiment were counted, in at least 3 matched experiments.
Invasion assay
Fluoroblok™ inserts with 8 μm membrane pores, 24 well plate format (BD Falcon – Ref. 351152) were coated with 50 μl Geltrex (Gibco – Life Technologies – Cat. Nr. A14132-02) diluted in RPMI or DMEM with 0.5% FCS (gradient) or with 10% FCS (random) to a final concentration of 1.2–1.8 mg/ml. Then, coated inserts were stored at 37°C for 1 h, to allow Geltrex to polymerize. In the lower chamber of the invasion well, 600 μl RPMI or DMEM supplemented with 10% FCS were added. 1E5 cells were resuspended in a volume of 200 μl of medium with 0.5% FCS (gradient) or 10% FCS (random) and subsequently were plated in the upper chamber of the invasion well. Plates were then stored at 37°C and allowed to invade for 24 h. Afterwards, the upper chamber was cleaned with a cotton swab and the inserts were analyzed on a Zeiss Axiovert 200M with a 10x objective. 4 images/well, corresponding to ¼ of the total area of the invasion insert, were analyzed in every condition. Cells were counted in every image with the “cell counter” plugin of ImageJ and each condition was compared to control cells.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the German Science Foundation (DFG) to HF and by grants from the Swiss Science Foundation (SNF) to HF and DFL.
References
- 1. Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends in Cell Biology 2006; 16:522-9. [DOI] [PubMed] [Google Scholar]
- 2. Hall A. Rho family GTPases. Biochem Soc Trans 2012; 40:1378-82. [DOI] [PubMed] [Google Scholar]
- 3. Etienne-Manneville S. Cdc42 - the centre of polarity. Journal of Cell Science 2004; 117:1291-300. [DOI] [PubMed] [Google Scholar]
- 4. Wang F, Herzmark P, Weiner OD, Srinivasan S, Servant G, Bourne HR. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Biol 2002; 4:513-8. [DOI] [PubMed] [Google Scholar]
- 5. Liu BP, Burridge K. Vav2 activates Rac1, Cdc42, and RhoA downstream from growth factor receptors but not beta1 integrins. Mol Cell Biol 2000; 20:7160-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3[beta] and adenomatous polyposis coli to control cell polarity. Nature 2003; 421:753-6. [DOI] [PubMed] [Google Scholar]
- 7. Michaelson D, Silletti J, Murphy G, D’Eustachio P, Rush M, Philips MR. Differential Localization of Rho Gtpases in Live Cells: Regulation by Hypervariable Regions and Rhogdi Binding. The Journal of Cell Biology 2001; 152:111-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Erickson JW, Zhang C-j, Kahn RA, Evans T, Cerione RA. Mammalian Cdc42 Is a Brefeldin A-sensitive Component of the Golgi Apparatus. Journal of Biological Chemistry 1996; 271:26850-4. [DOI] [PubMed] [Google Scholar]
- 9. Baschieri F, Confalonieri S, Bertalot G, Di Fiore PP, Dietmaier W, Leist M, et al. Spatial control of Cdc42 signalling by a GM130–RasGRF complex regulates polarity and tumorigenesis. Nat Commun 2014; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Farhan H, Rabouille C. Signalling to and from the secretory pathway. Journal of Cell Science 2011; 124:171-80. [DOI] [PubMed] [Google Scholar]
- 11. Yang G, Cynader MS. Regulation of protein trafficking: JNK3 at the Golgi complex. Cell Cycle 2014; 13:5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Millarte V, Farhan H. The Golgi in cell migration: regulation by signal transduction and its implications for cancer cell metastasis. ScientificWorldJournal 2012; 2012:498278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baschieri F, Farhan H. Crosstalk of small GTPases at the Golgi apparatus. Small GTPases 2012; 3:80-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCaffrey Luke M, Montalbano J, Mihai C, Macara Ian G. Loss of the Par3 Polarity Protein Promotes Breast Tumorigenesis and Metastasis. Cancer Cell 2012; 22:601-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xue B, Krishnamurthy K, Allred DC, Muthuswamy SK. Loss of Par3 promotes breast cancer metastasis by compromising cell–cell cohesion. Nat Cell Biol 2013; 15:189-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, et al. Deregulation of Scribble Promotes Mammary Tumorigenesis and Reveals a Role for Cell Polarity in Carcinoma. Cell 2008; 135:865-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weller SG, Capitani M, Cao H, Micaroni M, Luini A, Sallese M, et al. Src kinase regulates the integrity and function of the Golgi apparatus via activation of dynamin 2. Proc Natl Acad Sci U S A 2010; 107:5863-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bard F, Mazelin L, Péchoux-Longin C, Malhotra V, Jurdic P. Src Regulates Golgi Structure and KDEL Receptor-dependent Retrograde Transport to the Endoplasmic Reticulum. Journal of Biological Chemistry 2003; 278:46601-6. [DOI] [PubMed] [Google Scholar]
- 19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74. [DOI] [PubMed] [Google Scholar]
- 20. Qiao X, Roth I, Feraille E, Hasler U. Different effects of ZO-1, ZO-2 and ZO-3 silencing on kidney collecting duct principal cell proliferation and adhesion. Cell Cycle 2014; 13:3059-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bilder D. Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes & Development 2004; 18:1909-25. [DOI] [PubMed] [Google Scholar]
- 22. Gligorijevic B, Bergman A, Condeelis J. Multiparametric Classification Links Tumor Microenvironments with Tumor Cell Phenotype. PLoS Biol 2014; 12:e1001995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patsialou A, Bravo-Cordero JJ, Wang Y, Entenberg D, Liu H, Clarke M, et al. Intravital multiphoton imaging reveals multicellular streaming as a crucial component of in vivo cell migration in human breast tumors. Intravital 2013; 2:e25294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dang I, Gorelik R, Sousa-Blin C, Derivery E, Guerin C, Linkner J, et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature 2013; 503:281-4. [DOI] [PubMed] [Google Scholar]
- 25. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer cell 2006; 10:515-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma B, van Blitterswijk CA, Karperien M. A Wnt/β-catenin negative feedback loop inhibits interleukin-1–induced matrix metalloproteinase expression in human articular chondrocytes. Arthritis & Rheumatism 2012; 64:2589-600. [DOI] [PubMed] [Google Scholar]
- 27. Riaz M, Elstrodt F, Hollestelle A, Dehghan A, Klijn JG, Schutte M. Low-risk susceptibility alleles in 40 human breast cancer cell lines. BMC Cancer 2009; 9:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kao J, Salari K, Bocanegra M, Choi Y-L, Girard L, Gandhi J, et al. Molecular Profiling of Breast Cancer Cell Lines Defines Relevant Tumor Models and Provides a Resource for Cancer Gene Discovery. PLoS ONE 2009; 4:e6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weigelt B, Warne PH, Downward J. PIK3CA mutation, but not PTEN loss of function, determines the sensitivity of breast cancer cells to mTOR inhibitory drugs. Oncogene 2011; 30:3222-33. [DOI] [PubMed] [Google Scholar]
- 30. Krypuy M, Ahmed A, Etemadmoghadam D, Hyland S, Australian Ovarian Cancer Study G, deFazio A, et al. High resolution melting for mutation scanning of TP53 exons 5-8. BMC Cancer 2007; 7:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. The Journal of Clinical Investigation 2011; 121:2750-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 2008; 14:518-27. [DOI] [PubMed] [Google Scholar]
- 33. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012; 486:346-52. [DOI] [PMC free article] [PubMed] [Google Scholar]