

Abstract
Mammalian CST (CTC1-STN1-TEN1) is a telomere-associated complex that functions in telomere duplex replication and fill-in synthesis of the telomeric C-strand following telomerase action. CST also facilitates genome-wide replication recovery after HU-induced fork stalling by increasing origin firing. CTC1 and STN1 were originally isolated as a DNA polymerase α stimulatory factor. Here we explore how CST abundance affects recovery from drugs that cause different types of DNA damage and replication stress. We show that recovery from HU and aphidicolin induced replication stress is increased by CST over-expression. Elevated CST increases dNTP incorporation and origin firing after HU release and decreases the incidence of anaphase bridges and micronuclei after aphidicolin removal. While the frequency of origin firing after HU release is proportional to CST abundance, the number of cells entering S-phase to initiate replication is unchanged by CST overexpression or STN1 depletion. Instead the CST-related changes in origin firing take place in cells that were already in S-phase at the time of HU addition, indicating that CST modulates firing of late or dormant origins. CST abundance also influences cell viability after treatment with HU, aphidicolin, MMS and camptothecin. Viability is increased by elevated CST and decreased by STN1 depletion, indicating that endogenous CST levels are limiting. However, CST abundance does not affect viability after MMC treatment. Thus, CST facilitates recovery from many, but not all, forms of exogenous DNA damage. Overall our results suggest that CST is needed in stoichiometric amounts to facilitate re-initiation of DNA replication at repaired forks and/or dormant origins.
Keywords: CTC1, DNA repair, replication origin, DNA replication, STN1, telomere, TEN1
Abbreviations
- CldU
chlorodeoxyuridine
- CPT
camptothecin
- CST
CTC1-STN1-TEN1
- EdU
ethenyl deoxyuridine
- HU
hydroxyurea
- IdU
iododeoxyuridine
- MMC
mitomycin C
- MMS
methyl methanesulfonate
- MTS
multiple telomere signals
Introduction
Although genomes must be duplicated efficiently and with high fidelity, DNA replication is a complex process that is easily blocked by obstacles such as DNA damage and naturally occurring chromosomal structures. Failure to restart replication leads to collapse of the replication fork with formation of double-strand breaks, unwanted recombination intermediates and the risk of incomplete genome replication.1,2 To avoid such deleterious events, cells have evolved various mechanisms to deal with replication blocks. These include the use of additional proteins to aid passage of the replication fork,3 the ATR-mediated checkpoint pathway to help prevent fork collapse4 and the use of backup or dormant origins to ensure that replication forks can traverse all regions of the genome.5
Telomeres form a natural replication barrier due to their repetitive DNA sequence and chromatin structure.3,6 Consequently, efficient replication of the telomeric duplex requires the assistance of accessory factors such as helicases and nucleases in addition to the conventional replication machinery.3 Depletion of these accessory factors leads to defects in telomere structure and/or telomere loss. Recent studies have identified the mammalian CTC1-STN1-TEN1 (CST) complex as a key accessory factor that functions in several aspects of telomere replication.7-13
Mammalian CST resembles the Cdc13-Stn1-Ten1 complex from Saccharomyces cerevisiae (ScCST) in that the STN1 and TEN1 subunits are similar in structure, both complexes bind ssDNA, localize to telomeres and participate in telomere replication.7,14-20 During telomere replication, ScCST regulates telomerase-mediated elongation of the 3′ G-rich strand and coordinates subsequent fill-in synthesis of the complementary C-strand.21-24 ScCST also functions in telomere protection by preventing degradation of telomeric DNA by nucleases.25 Mammalian CST does not appear to participate in telomere protection but instead plays a wider role in DNA replication. During telomere replication, CST initially facilitates duplication of the telomere duplex DNA then later participates in C-strand fill-in following telomerase action.8-12 Unexpectedly, CST was also found to help resolve replication stress in non-telomeric regions after treatment with hydroxyurea (HU) to induce genome-wide replication fork stalling.11 CST-depleted cells exhibited less efficient restart of DNA synthesis after HU removal and a concomitant decrease in firing of new replication origins. Currently the mechanism(s) by which CST promotes replication at telomeres and elsewhere in the genome is unknown, but since CTC1 and STN1 were originally identified as factors that enhance DNA polymerase α-primase processivity and affinity for ssDNA templates, modulation of pol α activity seems a likely pathway.26,27
CST is essential for human health as loss of function mutations in CTC1 cause the neurological disorder Coats plus and can also result in the telomere maintenance disorder dyskeratosis congenita.28,29 Interestingly, several observations point to CTC1/CST misregulation in some cancers. First, CTC1 gene copy number is frequently increased in canine and human osteosarcoma.30 Second, the Oncomine database indicates anomalous expression of CTC1, STN1 and TEN1 mRNA in many tumors and upregulation of gene expression is particularly striking in ductal breast carcinomas.31-35 Abnormal expression of DNA repair factors is common in many forms of cancer and can have implications for patient prognosis and treatment.36-38 Upregulation of certain repair proteins can decrease survival by increasing the risk of metastasis and causing resistance to certain forms of chemotherapy.39-41 Down regulation of DNA repair proteins is a known risk factor for tumor formation.38,42 Thus, the presence of abnormal CST levels in certain cancers could help guide treatment and/or be an indicator of patient survival.
With this possibility in mind, we have now examined how CST levels affect the cellular response to a variety of DNA-damaging agents. We have used CST overexpression and CST/STN1 depletion as complementary approaches to gain information about the role of CST in recovery from replication stress and hence the potential impact of CST misregulation in cancer patients. Our results indicate that CST helps cells survive multiple forms of DNA damage and that endogenous levels of CST are limiting for recovery.
Results
Establishment of CST over-expressing cells
To examine the effect of CST over-expression on telomere replication and genome-wide replication rescue, we established a HeLa cell line that over-expressed the 3 subunit CTC1-STN1-TEN1 complex (CST O/E) (Fig. 1). TEN1 was under control of a tetracycline-inducible promoter while HA-tagged CTC1 and FLAG-tagged STN1 were expressed constitutively. The parental cells expressed the Tet-On transactivator (HeLa Tet-On). We also made cell lines that overexpressed TEN1 or TEN1 + STN1 because prior knockdown experiments hinted that TEN1 might function independently of STN1 and CTC1.13 We did not over-express TEN1 + CTC1 because CTC1 depends on STN1 for stability.9,27,43 Western blots with STN1 or TEN1 antibody indicated a ∼16-fold increase in STN1 and an ∼8 fold increase in TEN1 relative to control cells (Fig. 1A). Quantification of CTC1 protein was not possible due to lack of a suitable antibody however real time RT-PCR indicated a ∼40 fold increase in CTC1 mRNA (Fig. 1B). The rise in protein level was likely less than the increase in mRNA as the increase in STN1 and TEN1 mRNA (∼70× and ∼14×) significantly exceeded that of the protein.
Figure 1.
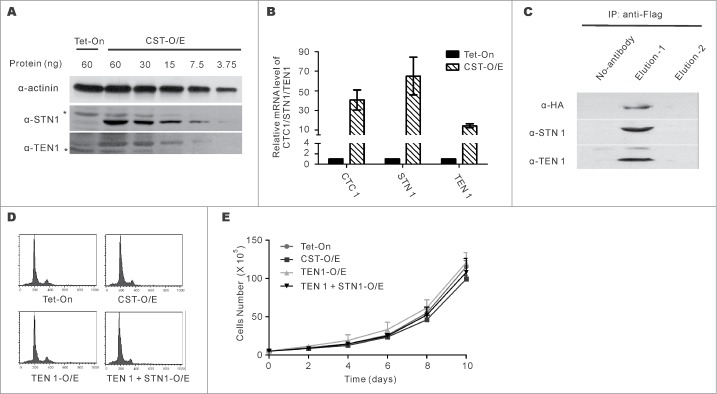
Characterizing the CST overexpression cell line (CST-O/E). (A) Western blots with STN1 or TEN1 antibody showing levels of over-expression in CST-OE cells relative to Tet-On control. Amount of cell lysate (ng protein) is shown above each lane. *indicates cross-reacting band. (B) RT-qPCR of CTC1, STN1 and TEN1 mRNA levels. Levels are relative to control Tet-On cells with normalization to GAPDH (mean ± SEM, n = 3 experiments). (C) Western blot of FLAG-STN1 immunoprecipitate showing co-purification of HA-CTC1 and TEN1. (D) Analysis of DNA content by FACS shows normal cell cycle profile in CST, TEN1 and TEN1 + STN1 over-expressing cells. (E) Representative growth curves for the indicated cell lines (mean ± SEM, n = 3 experiments). Cells were counted in triplicate for each time point.
We confirmed by co-immunoprecipitation that HA-tagged CTC1 and FLAG-tagged STN1 are competent for CST complex formation. FLAG-STN1 was immunoprecipitated from CST-O/E whole cell extracts using FLAG antibody-coupled beads and eluted with FLAG peptide. Western blots with antibody to HA and TEN1 indicated that HA-CTC1 and TEN1 co-purified with FLAG-STN1 (Fig. 1C). We next examined whether growth rate or cell cycle profile were affected by CST overexpression. No changes were observed in cells over-expressing TEN1 alone, STN1 + TEN1 or all 3 CST subunits (Fig. 1D and E).
CST over-expression does not affect telomere maintenance
To address whether increased CST affects telomere replication, we first examined G-overhang status. Relative overhang amount was assessed by in-gel hybridization of probe to telomeric restriction fragments under non-denaturing conditions.12 Gels were then denatured and re-hybridized with the same probe to allow quantification of overhang signal relative to total telomere signal (Fig. S2A and B). This analysis revealed no significant change in overhang amount in CST O/E cells relative to the control cells. Thus, CST overexpression does not appear to affect fill-in synthesis of the C-strand following G-strand elongation by telomerase. An additional role for CST is in facilitating DNA replication through the telomere duplex.10,11 This role is particularly important in cells with long telomeres where CST loss of function manifests as multiple telomere signals (MTS) on an individual chromosome end when telomeres are visualized by FISH.12 We looked for MTS in metaphase spreads from Tet-On and CST O/E cells but the background level was very low and there was no apparent change after CST over-expression (data not shown). This observation fits with the Tet-On cells having short telomeres (Fig. S2C). Likewise, CST overexpression caused no measurable change in chromosome fusions, telomere loss or any other telomere abnormality (data not shown).
It has been reported that CST may function in telomere length regulation in HT1080 cells by suppressing telomerase action.16 We therefore examined whether CST over-expression affects telomere length. Control Tet-On and CST-O/E cells were cultured for 35–50 PD and genomic DNA isolated at intervals for telomere length analysis. CST levels were monitored throughout the culture period and cells were re-sorted when necessary to maintain high levels of expression (Fig. S1). Southern blot analysis of telomeric restriction fragments revealed that CST overexpression has negligible effect on telomere length as the median length in the control and CST-O/E cells was maintained at ∼3.7 kb throughout the time course (Fig. S2C). Overall our results indicate that the CST complex is not a limiting factor for telomere maintenance in HeLa cells.
Elevated CST enhances recovery from replication stress
Given the genome-wide role for CST in recovery from replication stress, we asked whether CST over-expression could enhance recovery from stress caused by nucleotide depletion or partial polymerase inhibition. To examine recovery of DNA synthesis after HU-induced nucleotide depletion, we treated control Tet-On cells, TEN1, TEN1 + STN1, or CST-O/E cells with HU for 2 hours to stall replication.11 The HU was then removed and EdU was added to label cells that re-initiated replication. Actively replicating cells were identified by immunofluorescence and the EdU signal per nucleus was quantified11 (Fig. 2A and B). In the absence of HU treatment, the percent of EdU positive cells and the levels of EdU incorporation were similar in all 4 cell lines indicating that there was no difference in the number of cells in S-phase or in the rates of replication. This was also true for the control cells and the TEN1 or TEN1 + STN1 over-expressing cells after release from HU. In contrast, the CST-O/E cells exhibited a ∼40% increase in EdU uptake per nucleus. Thus, recovery of DNA synthesis after HU treatment is enhanced by an increased level of the 3 subunit CST complex but not by elevated TEN1 or TEN1 + STN1.
Figure 2.
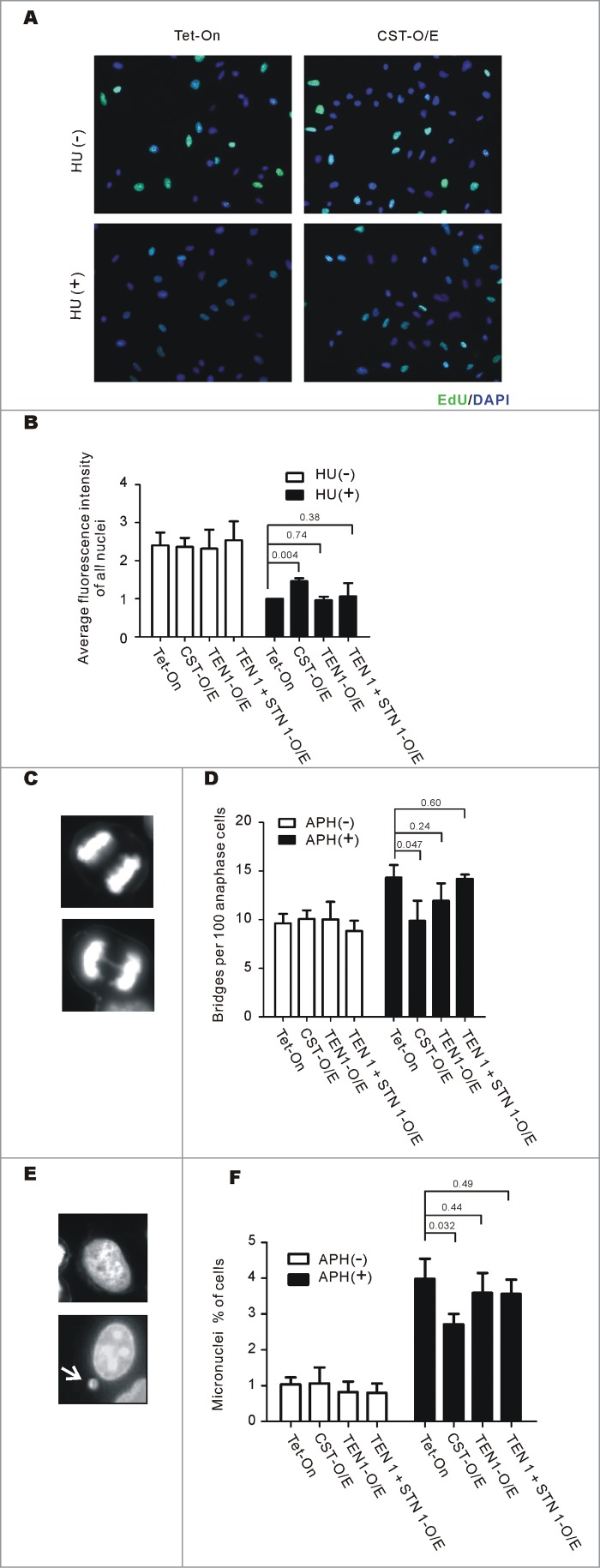
CST overexpression rescues the effects of replication stress. (A and B) Replication restart after release from HU. CST, TEN1, STN1 + TEN1 over-expressing cells were treated with HU for 2 h, released and then labeled with EdU for 30 min. (A) Images showing EdU uptake. Green, EdU; Blue, DAPI (B) Quantification of EdU uptake. Levels are relative to EdU incorporation by control Tet-On cells after release from HU (mean ± SEM, n = 3 experiments, p-values are indicated). (C-F) Cells were treated with 0.2 mM aphidicolin for 16 h (C and D) or 10 h (E and F) to induce formation of anaphase bridges and micronuclei. (C) Representative images of cells with (bottom) or without (top) anaphase bridges. (D) Quantification of the percentage of anaphase cells with bridges (mean ± SEM, n = 3 experiments, p-values are indicated). (E) Images of cells with (bottom) or without (top) micronuclei. The arrow points to a micronucleus. (F) Quantification of the percent of cells with micronuclei (mean ± SEM, n = 3 experiments, p-values are indicated).
We previously observed that partial depletion of CTC1, STN1 or TEN1 leads to an increase in DAPI-stained anaphase bridges and micronuclei without a concomitant increase in telomere fusions. As anaphase bridges and micronuclei can arise from unresolved replication intermediates,44-46 our finding suggested a role for CST in the resolution of replication stress at natural replication barriers.11,13 To follow up on this possibility, we examined whether CST overexpression could prevent the formation of anaphase bridges and micronuclei after stalling of replication forks with low doses of the polymerase inhibitor aphidicolin. Control cells, CST-O/E cells and cells over-expressing TEN1 or TEN1 + STN1 were grown with or without aphidicolin and then monitored for the percent of anaphase cells displaying anaphase bridges and frequency with which cells harbored micronuclei (Fig. 2C–F). Although the HeLa Tet-On cell line displayed a high basal level of anaphase bridges, as expected the aphidicolin treatment caused a further increase. The basal level of anaphase bridges in the 3 over-expression cell lines was similar to the control and like the control cells, the TEN1 and TEN1 + STN1 cells displayed an increase in bridges after aphidicolin treatment. However, this increase in anaphase bridges was completely rescued by CST overexpression. The background level of micronuclei was also similar in the 4 cell lines and the control Tet-On, TEN1 and TEN1 + STN1 cells showed a similar increase in micronuclei in response to aphidicolin. However, this was partially rescued by over-expression of the 3 subunit CST complex. Since the anaphase bridges and micronuclei observed after aphidicolin treatment are not generally caused by telomere fusions or a specific failure in telomere replication,10,11,47 our results again indicate that CST-overexpression enhances resolution of replication stress genome-wide. Overall, our findings indicate that recovery from HU and aphidicolin induced replication fork stalling depends on the 3 subunit CST complex and in HeLa cells the endogenous level of CST is limiting for this recovery.
Elevated CST increases origin firing after HU-induced fork stalling
We previously demonstrated that STN1 depletion impedes recovery from HU treatment by causing a decrease in origin firing rather than by affecting fork restart.11 To explore whether CST over-expression enhances recovery from HU treatment through the same mechanism, we used DNA fiber analysis to directly examine DNA replication events at the molecular level (Fig. 3). Control Tet-On and CST-O/E cells were labeled with IdU for 20 min, replication was then stalled by addition of HU for 2 hours. The cells were released into media containing CIdU for 25 min, harvested, lysed on microscope slides and the DNA fibers spread by hydrodynamic flow as previously described.11 The labeled DNA fibers were stained with antibodies to IdU and CIdU, visualized by confocal microscopy and the replication events quantified (Fig. 3B–D). Examination of the red-only (IdU) tracks which correspond to fork termination and fork stalling events, revealed a similar increase after HU treatment in the control and CST-O/E cells indicating that elevated CST did not affect the frequency with which stalled replication forks were able to re-start. However, CST overexpression caused a significant increase in green-only (CldU) tracks which correspond to origin firing after HU release. This increase in new origin firing was accompanied by a modest but not statistically significant decline in elongating forks (red-green tracks). Thus, it appears that the ability of cells to recover from HU-induced fork stalling is directly related to the role played by CST in enhancing origin firing.
Figure 3.
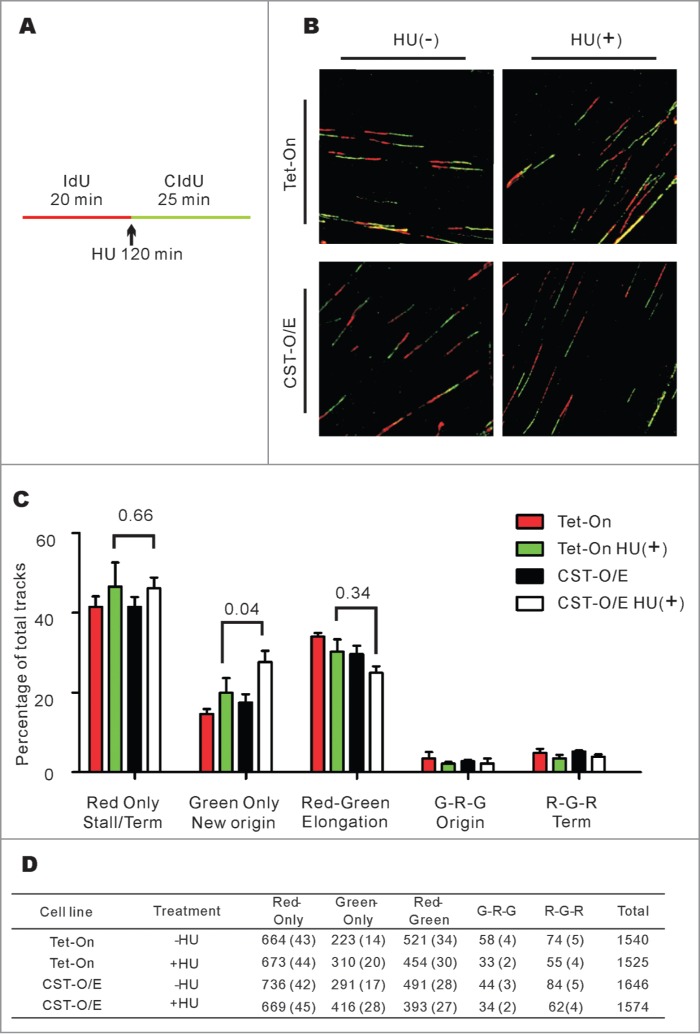
CST over-expression leads to an increase in new origin firing following release from HU-induced fork stalling. (A) Schematic showing the timing of HU treatment relative to IdU and CldU labeling. (B) Representative images of DNA fibers. Red, IdU; green, CldU. (C) Graph showing the percentage of the indicated types of track (Mean ± SD, n = 3 experiments, p-values are indicated). (D) Total number of tracks scored, number in brackets indicates the percentage of total tracks scored G, Green; R, Red.
CST promotes firing of late or dormant origins
Although our DNA fiber studies demonstrated that CST levels determine the frequency of origin firing after release from HU, it remained possible that the altered origin firing merely reflects a cell cycle effect of CST depletion11 or over-expression (this publication). Since HU blocks the cell cycle at the G1/S boundary, CST levels might determine the rate at which cells enter S-phase after HU removal and hence how quickly they initiate firing of early origins.
To assess whether CST levels have such an effect on cell cycle entry, we devised an approach based on EdU and PCNA labeling to differentiate between cells newly entering S-phase after HU removal versus those that had entered S-phase prior to HU addition (Fig. 4A). We then compared the number of control cells vs. CST-overexpressing cells entering S-phase after HU release. We also performed the experiment with the STN1-depleted cell line (shSTN1–7) used in our previous analysis of origin firing.11 For the latter experiments, we included control cells expressing a non-targeting shRNA (shNT) or STN1 shRNA plus a rescuing FLAG-tagged sh-resistant STN1 allele (shSTN1–7 Res). All cell lines were characterized previously11 and STN1 protein was barely detectable in the shSTN1 cells (Fig. S3).
Figure 4.
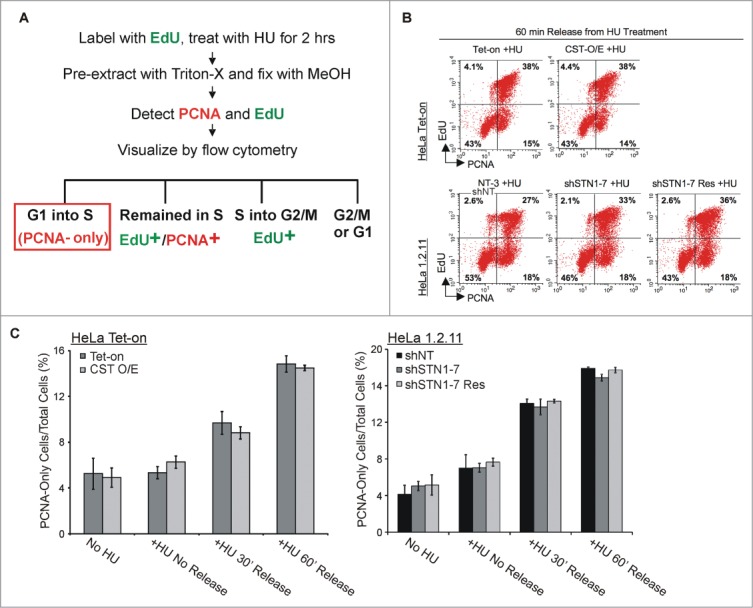
Alteration in CST abundance does not change cell cycle entry following HU treatment. (A) Schematic showing the experimental design. HeLa Tet-On or HeLa 1.2.11 cells were labeled with EdU, treated with HU for 2 h and then released for 0, 30 or 60 min. Cells not treated with HU were used as a control. EdU and PCNA were then detected to compare the fraction of cells entering S-phase (PCNA-only) relative to cells already in S-phase (PCNA+/EdU+) or just exiting (EdU+) S-phase. (B) Examples of flow cytometry data after 60 min release from HU treatment showing the percent of cells labeled with EdU and/or PCNA. Top: Control and CST overexpressing cell lines (HeLa Tet-On and CST-O/E); Bottom: STN1 depleted (shSTN1–7 clone) and control sh non-target (shNT) and shSTN1 rescue (shSTN1–7 Res) HeLa 1.2.11 cells. Quadrants were created using control cells without EdU or PCNA staining. (C) Graphical representation of the percent of cells entering S-phase (PCNA-only) (mean ± SEM, n ≥ 4 independent experiments).
To distinguish between the cells entering S-phase from those that were already in S-phase, cells were pre-labeled with EdU for 30 min prior to HU addition. The presence of chromatin-bound PCNA was then used as a marker for cells that were in S-phase 30 or 60 min after release from HU. Cells with EdU and/or PCNA staining were detected by flow cytometry (Fig. 4). Cells with PCNA labeling but no EdU labeling represented the fraction of cells that had accumulated at the G1/S boundary during the HU treatment and then entered S-phase when the HU was removed (Fig. 4A and B). As shown in Figure 4B–D, a similar fraction of cells entered S-phase (PCNA-only) after the release from HU in the control, STN1-depleted and CST over-expressing cell lines. This finding supports previous analyses indicating that our shSTN1 and CST-OE cell lines show a normal cell cycle distribution (Fig. 1D and11). Hence, the change in origin firing observed in response to altered levels of CST/STN1 cannot be explained merely by differences in the number of cells entering S-phase to initiate DNA replication. Instead, it implies that CST affects origins in cells that were already in S-phase at the time of HU addition. In which case, the affected origins would be late firing origins or dormant origins. The above conclusion is supported by our DNA fiber data11 (Fig. 3) where essentially all the fibers used to score new origins (green, CldU-labeled tracks) lay adjacent to fibers with red/green tracks indicating elongating forks. Regions of the slide that had only green tracks were excluded from the analysis as they likely arose from a cell entering S-phase which only incorporated CldU and thus were unsuitable for analysis of fork restart. Thus, since the red/green tracks originated from cells that were in S-phase during the IdU labeling period, the adjacent scored origins were mostly from cells that had entered S-phase prior to HU treatment, again indicating that they correspond to dormant or late firing origins. Given that recovery from HU treatment is known to increase firing of dormant and late origins,48,49 our finding strongly support a role for CST in modulating the firing of these origins in response to replication stress.
CST levels determine sensitivity to a variety of DNA damaging agents
Although HU treatment was used to demonstrate the genome-wide role of CST in recovery from replication stress,11,13 the long term effect of HU on cells with altered levels of CST was not determined. We therefore examined the viability of both CST over-expressing cells and STN1 depleted cells after 4–20 h HU treatment. Cells were treated with 2 mM HU to fully inhibit DNA replication,11 then returned to fresh medium for 24 h prior to analysis by MTT assay (Fig. 5A). All the cell lines tested showed a decline in viability at the longer treatment times which is consistent with the occurrence of replication fork collapse after prolonged fork stalling.50 However, this decline was markedly faster in the STN1-depleted cells and it occurred even after short periods of HU treatment. In contrast, the CST-O/E cells showed a small but consistent increase in viability. These results indicate that CST is important for cells to survive prolonged replication fork stalling.
Figure 5.
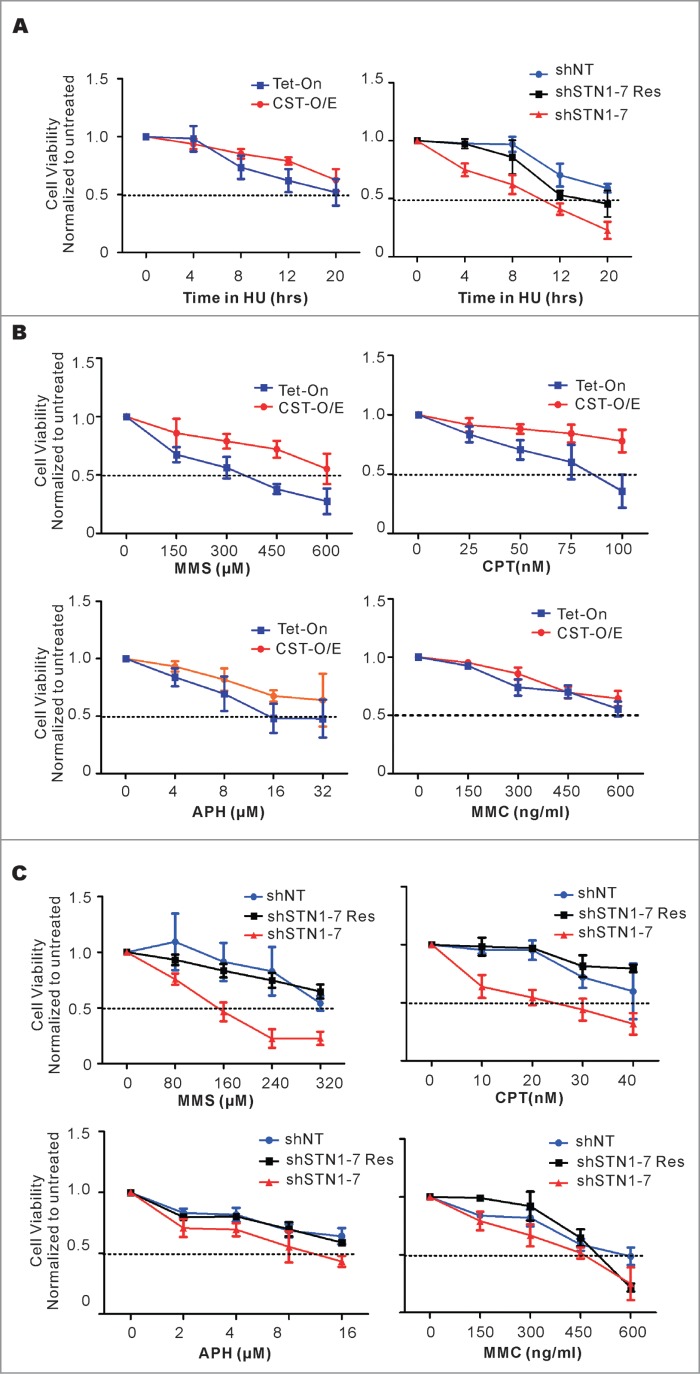
CST rescues cells from the cytotoxic effects of diverse DNA-damaging agents. (A) Cells were treated with 2 mM HU for the indicated times, released for 24 h and relative cell number determined by MTT assay. Left panel, results from CST-O/E and Tet-On control cells. Right panel, results from shSTN1 cells and shNT or shSTN1-Res control cells. (B and C) Cells were treated with the indicated concentrations of methyl methanesulfonate (MMS) for 8 h, camptothecin (CPT) for 16 h, aphidicolin (APH) for 8 h or mitomycin C (MMC) for 12 h, allowed to recover for 24 h and viability/proliferation measured by MTT assay. (B) Effect of drugs on over-expressing cells (CST-O/E) or control Tet-On cells. (C) Effect of drugs on shSTN1 or control shNT and shSTN1-Res cells. Each time point was assayed in triplicate and the data are shown as the mean ± S.D from 3 or 4 independent experiments. For each cell line, the value of the untreated sample was set at 1.
To further explore the importance of CST in recovery from exogenous DNA damage, we asked whether CST helps cells survive treatment with drugs that damage DNA through different mechanisms. Both CST-O/E and shSTN1 cells were treated with increasing concentrations of the alkylating agent methyl methanesufonate (MMS), the topoisomerase inhibitor camptothecin (CPT), the DNA polymerase inhibitor aphidicolin and the DNA cross-linker mitomycin C (MMC) (Fig. 5B and C). The cells were allowed to recover for 24 h and then analyzed for viability by MTT assay. The results were striking as the STN1 depleted cells were considerably more sensitive to both MMS and CPT than the control cells while the CST over-expressing cells were less sensitive than the control. The resistance of the CST-O/E cells to MMS and CPT was confirmed by colony formation assay (data not shown). The effects of aphidicolin were less dramatic but the shSTN1 cells again showed lower viability and the CST-O/E cells had higher viability than the control cells. In contrast, the response to MMC was unaffected by CST overexpression or depletion. Thus, CST facilitates recovery from many, but not all, forms of exogenous DNA damage.
Discussion
Past analysis of the genome-wide roles of mammalian CST has focused on the effect of CST depletion on recovery from HU-induced replication fork stalling.11,13 We have now performed a broader analysis to assess how changes in CST abundance affect recovery both from HU and a series of drugs that cause different types of DNA damage and thus elicit a variety of repair and replication rescue pathways. We show that the degree of recovery from these agents is generally proportional to the amount of CST in a cell as STN1 depletion renders cells more sensitive to drug treatment while over-expression of the 3 subunit complex confers resistance. We therefore conclude that endogenous CST levels are limiting for its non telomeric role(s). In contrast, endogenous CST levels are sufficient for telomere replication. The opposing response to CST/STN1 depletion versus overexpression following genome-wide DNA damage is striking and reminiscent of the effects of RPA exhaustion and excess,51 suggesting that CST may also be needed in stoichiometric amounts to resolve replication stress.
Following HU treatment, CST abundance affects replication recovery by modulating the frequency of origin firing. STN1 depletion decreases new origin firing11 while CST over-expression increases origin firing relative to control cells (Fig. 3). Moreover, most origins impacted by CST reside in cells that were in S-phase prior to HU addition, indicating that CST promotes the firing of dormant or late origins. The decrease in new origin firing after STN1 depletion contrasts with the increase in origin firing seen after depletion of the RTEL or BLM helicases.52,53 These helicases act at the replication fork to help remodel stalled forks and resolve recombination intermediates. Thus, the inverse relationship between helicase level and frequency of origin firing is likely an indirect effect of ATR signaling from unresolved structures at the fork, which can stimulate firing of dormant origins in the same replicon.49 Our finding that origin firing is proportional to CST abundance suggests that CST may have a more direct effect on origin activation then RTEL or BLM. Given the ability of CST to stimulate DNA pol α-primase activity, one possibility is that CST responds to replication stress by aiding in polymerase recruitment or activation at late or dormant origins.
The finding that CST levels impact the viability of cells following treatment with CPT, MMS, HU and aphidicolon but not MMC is intriguing given the range of lesion caused by these drugs and the varied degree of overlap in the subsequent repair pathways. MMS mostly generates N7-methylguanine and N3-methyladenine which are removed by base excision repair.54 However, during DNA replication both lesions block fork elongation and the stalled forks are thought to collapse into DSB which are repaired by homologous recombination (HR).55 CPT inhibits topoisomerase I (TopI) resulting in formation of TopI-DNA cleave complexes.56 These complexes block the replication fork and, as with MMS, the stalled forks are converted into DSB which are repaired by HR.57 While we cannot rule out a replication-independent role for CST in recovery from MMS damage, the concentration of CPT used in this study primarily causes cytotoxicity through inhibition of DNA replication.56 Moreover, HU and aphidicolin act specifically at the level of DNA polymerase through nucleotide depletion and polymerase inhibition respectively. Thus, it seems likely that the effects of CST abundance on viability after treatment with these drugs relates to its role in replication.
The severity of the replication fork damage resulting from MMS and CPT-induced lesions contrasts with the relatively minimal damage caused by short term HU or aphidicolin treatment. The rapid resumption of replication after removal of HU or aphidicolin indicates that the replisome must be largely intact.11,52 In contrast, it would have to be re-assembled following repair of a MMS or CPT-induced DSB, raising the possibility that CST helps establish DNA pol α at repaired forks in addition to acting at late firing or domant origins.
It is unclear why recovery from MMC is not affected by CST abundance given that the resulting intrastrand cross-links (ICL) completely block the replication fork and their removal also involves the formation of a DSB and repair by HR. However, ICL repair is a complex process that involves the Fanconi anemia signaling pathway and a number of additional steps beyond those required to resolve MMS or CPT induced lesions.58 Thus, it is possible that the mechanism used to re-establish replication during ICL repair is also different.
A common feature of the drugs we used to test for response to CST levels is that they all elicit an ATR-mediated S-phase checkpoint.4,59 Interestingly while MMC elicits the checkpoint, ATR activation is via the Fanconi anemia core complex rather than the canonical pathway involving Rad17.59-61 Thus, our findings raise the possibility that the action of CST is somehow linked to a specific aspect of ATR signaling. We previously showed that STN1 depleted cells exhibit a normal ATR response to HU treatment as the level of Chk1 phosphorylation is similar to that of control cells as is the rate of Chk1 dephosphorylation and loss of RPA foci after HU release.11 We therefore question whether CST might help resolve some aspect of the checkpoint reversal. Interestingly, the inhibition of origin firing by ATR/Chk1 involves phosphorylation of pre-initiation complex components.62,63 Thus, one role for CST might be to restore firing competency to modified pre-initiation complexes.
Regardless of the actual mechanism of CST action, our work shows that CST aids in recovery from a wide variety of DNA damaging agents. The relationship between CST abundance and cellular sensitivity to these agents is of direct medical relevance because their derivatives and other DNA damaging agents are commonly used in therapy. Functional CST levels may vary with disease state and thus render therapy more or less effective. Since Coats plus patients have partially inactive CST,64 treatment with DNA damaging agents is likely to cause elevated damage and may also result in stem cell depletion due to telomere loss. Conversely, the tumors of some cancer patients may have elevated CST and hence be more resistant to IR and certain forms of chemotherapy.30-35,65 Once antibodies suitable for immunohistochemistry become available, it will important to screen tumor samples to determine the frequency and level of CST expression in different types of tumor.
Materials and Methods
Cell culture and establishment of CST over-expressing cells
HeLa Tet-On cells (Clontech), HeLa 1.2.11 and H1299 cells were grown in RPMI with 10% FBS, antibiotics, and glutamine. For stable TEN1 expression, HeLa Tet-On cells were transfected with pTRE2-TEN1 plasmid, selected with 1 μg/ml hygromycin and single colonies isolated. TEN1 mRNA expression was confirmed by real-time RT-PCR (RT-qPCR) after 24 h induction with 1 ug/ml doxycycline. To create a stable cell line over-expressing all 3 CST subunits, a single clone showing inducible TEN1 expression was infected sequentially with retroviruses harboring pMIEG-FlagSTN1 and pMIT HA-CTC1. The viruses encoded GFP or Thy1.1 downstream of the STN1 or CTC1 genes. Cells expressing GFP or Thy 1.1 were isolated by FACS. Thy 1.1 detection was with APC-conjugated antibody. The CST over-expressing cells were cultured periodically in hygromycin to maintain TEN1 expression. CTC1 and STN1 levels gradually declined after prolonged culture due to lack of selective pressure so cells with high CTC1 and STN1 expression were re-isolated every ∼20 PD. Re-isolation was performed by flow cytometry based on GFP or Thy1.1 expression (Fig. S1).
Antibodies and Western blots
For TEN1 and STN1 detection, cells were lysed in Triton lysis buffer (50 mM Tris pH 7.2, 150 mM NaCl, 1.5 mM MgCl2, 1 mM DTT, 0.5 mM PMSF and 0.1% Triton X-100), 30 μg protein was separated by SDS-PAGE and transferred to nitrocellulose membrane. Membranes were blocked with 1% fish-gelatin and incubated with antibody to actinin (Santa Cruz; 1:50,000), HA (Cell Signaling 1:1000), Flag M2 (Sigma 1:1000), Histone H3 (Cell Signaling 1:2,500), TEN1 (1:500),13 and STN1 (1:1000)11 overnight then incubated with HRP-conjugated secondary antibody (Thermo; 1:500) for 30 min and developed using ECL Prime (GE Healthcare). Dilution series were performed to confirm detection was in the linear range and bands were quantified using Image J.
Co-immunoprecipitation
Cell lysates were prepared in NP-40 lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF and 0.5% NP-40) supplemented with protease inhibitor cocktail (Sigma). Immunoprecipitation was performed with anti-Flag M2 affinity gel (Sigma) with incubation overnight at 4 °C. Beads were washed 3 times with NP-40 lysis buffer, bound proteins eluted with Flag peptide (200 μg/ml) and analyzed by Western blot.
Telomere length and G-overhang analysis
Genomic DNA was isolated by proteinase K digestion and high salt precipitation.12 For telomere length determination, DNA was digested with HinfI and MspI overnight then restriction fragments were separated in 1% agarose gels. In-gel hybridization was performed using a (TA2C3)4 probe after denaturation with NaOH. For G-overhang analysis, control samples were treated with Exo1, the DNA was then digested with EcoRI and samples were separated briefly in 1% agarose gels to keep the telomeric restriction fragments in a tight band. In-gel hybridization was performed under native conditions using (TA2C3)4 probe, the DNA was then denatured and the gel rehybridized with the same probe. For each lane, the Exo1-resistant signal was subtracted from the untreated non-denatured signal and the resulting G-overhang signal was normalized for loading using the signal from the denatured sample.
Replication restart
Cells were plated onto coverslips, grown overnight to ∼30% confluency and then treated with 2 mM HU for 2 h as previously described.11 Cells were washed 3 times to remove the HU and media with 50 μM EdU (Invitrogen) was added for 30 min. The coverslips were then fixed with MeOH at −20°C for 10 min, processed using the Click-iT EdU AlexaFluor 488 Imaging Kit (Invitrogen), and stained with 0.2 μg/ml DAPI. The intensity of staining within individual nuclei was quantified using Image J software.
Anaphase bridges and micronuclei
Cells plated on coverslips were treated with 0.2 mM aphidicolin for 16 h, washed with PBS and treated with 50 ng/ml nocodazole for 3 hours. They were again washed with PBS and left in fresh media for 30–90 min prior to fixation in 3% formaldehyde for 10 min and staining with DAPI. To count micronuclei, cells were fixed directly after 10 hours aphidicolin treatment.
DNA fiber analysis
Cells were grown overnight and then labeled with 50 μM IdU (Sigma) for 20 min. The control cells (−HU) were then washed and labeled with 100 μM CldU (Sigma) for 25 min. The remaining cells were treated with 2 mM HU for 2 h, washed and released into media with 100 μM CldU for 30 min. The cells were harvested, used to prepare DNA fibers and the fibers were stained as previously described.11 The fibers were visualized with a Zeiss LSM710 confocal microscope at ×630 magnification. At least 150 fibers and 5 images were scored for each independent experiment. Scoring of fibers was performed using CASA software described previously.11
Analysis of S-phase entry
Cells were labeled with 50 μM EdU for 20 min, washed with PBS and treated with 2 mM HU for 2 h. Cells were released from HU and cultured for an additional 30 or 60 min in fresh media. Control cells were collected immediately after EdU labeling (+EdU) or after HU treatment (+HU). Following collection, the cells were washed once with 5 ml of PBS, pre-extracted with 0.5 ml of permeablization buffer (0.5% TritonX-100, 0.2 μg/ml EDTA, 1% BSA in 1× PBS) for 15 min and then immediately fixed with 5 ml of methanol at −20°C for 10 min. 5 ml of 1% BSA in PBS + 0.1% Tween-20 (PBST) was added and the cells were pelleted at 1000 × g for 5 min. Cells were resuspended in 1% BSA in 1× PBST and stored overnight at 4°C. Cells were incubated with PCNA antibody (PC-10, Santa Cruz) for 1 hr, washed with 1% BSA in 1× PBST and incubated with Alexa-647 conjugated secondary antibody (Invitrogen) for 30 min. EdU was then detected by click chemistry with Alexa-Fluor 488-Azide, as previously described.11 Following one wash with 1% BSA in 1× PBST, cells were treated with RNase for 15 min, the DNA stained with 7-AAD and the cells analyzed with a FACSCalibur (BD Biosciences) to determine the number of PCNA and/or EdU positive cells.
MTT assay
To monitor cell viability and proliferation, the tetrazolium based MTT colorimetric assay was performed as described66 with the following modifications. Cells were grown overnight in 24 well plates then treated with various drugs for the indicated time and released into growth medium without drugs for an additional 24 h. The culture medium was then replaced by 1 mg/ml MTT containing DMEM and left for 40 min at 37°C. The medium was removed and DMSO was added and left for 15 min with shaking at room temperature to dissolve the formazan crystals. The signal intensity was then measured with a multi-well scanning spectrophotometer (Synergy MX, BioTek) at 570 nm.
Statistical Analysis
The student's 2-tailed unpaired t-test was used to determine statistical significance and the resulting p-values are indicated in the figures.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Cyrus Vaziri for helpful discussions and reagents and Paul Andreassen for helpful comments. We thank Mary Chaiken for generation of constructs, the CCHMC Viral vector core for supplying lentivirus, the Cancer Biology microscopy core and Brigit Ehmer for help with confocal microscopy and cell sorting.
Funding
This work was supported by the National Institutes of Health grants GM041803 to CMP, F32 GM097833 and K99 GM104409 to JAS.
Supplemental Material
Supplemental data for this article can be found on the publisher's website.
References
- 1. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014; 16:2-9; PMID: 24366029; http://dx.doi.org/ 10.1038/ncb2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 2010; 11:208-19; PMID:20177396; http://dx.doi.org/ 10.1038/nrm2852 [DOI] [PubMed] [Google Scholar]
- 3. Leon-Ortiz AM, Svendsen J, Boulton SJ. Metabolism of DNA secondary structures at the eukaryotic replication fork. DNA Repair (Amst) 2014; 19:152-62; PMID:24815912; http://dx.doi.org/ 10.1016/j.dnarep.2014.03.016 [DOI] [PubMed] [Google Scholar]
- 4. Jossen R, Bermejo R. The DNA damage checkpoint response to replication stress: a game of forks. Front Genet 2013; 4:26; PMID:23493417; http://dx.doi.org/ 10.3389/fgene.2013.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blow JJ, Ge XQ, Jackson DA. How dormant origins promote complete genome replication. Trends Biochem Sci 2011; 36:405-14; PMID:21641805; http://dx.doi.org/ 10.1016/j.tibs.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Paeschke K, McDonald KR, Zakian VA. Telomeres: structures in need of unwinding. FEBS Lett 2010; 584:3760-72; PMID:20637196; http://dx.doi.org/ 10.1016/j.febslet.2010.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S, Saito M, Ishikawa F. RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell 2009; 36:193-206; PMID:19854130; http://dx.doi.org/ 10.1016/j.molcel.2009.08.009 [DOI] [PubMed] [Google Scholar]
- 8. Surovtseva YV, Churikov D, Boltz KA, Song X, Lamb JC, Warrington R, Leehy K, Heacock M, Price CM, Shippen DE. Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol Cell 2009; 36:207-18; PMID:19854131; http://dx.doi.org/ 10.1016/j.molcel.2009.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gu P, Min JN, Wang Y, Huang C, Peng T, Chai W, Chang S. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J 2012; 31:2309-21; PMID:22531781; http://dx.doi.org/ 10.1038/emboj.2012.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang C, Dai X, Chai W. Human Stn1 protects telomere integrity by promoting efficient lagging-strand synthesis at telomeres and mediating C-strand fill-in. Cell Res 2012; 22:1681-95; PMID:22964711; http://dx.doi.org/ 10.1038/cr.2012.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stewart JA, Wang F, Chaiken MF, Kasbek C, Chastain PD, 2nd, Wright WE, Price CM. Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J 2012; 31:3537-49; PMID:22863775; http://dx.doi.org/ 10.1038/emboj.2012.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang F, Stewart JA, Kasbek C, Zhao Y, Wright WE, Price CM. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep 2012; 2:1096-103; PMID:23142664; http://dx.doi.org/ 10.1016/j.celrep.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kasbek C, Wang F, Price CM. Human TEN1 maintains telomere integrity and functions in genome-wide replication restart. J Biol Chem 2013; 288:30139-50; PMID:24025336; http://dx.doi.org/ 10.1074/jbc.M113.493478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giraud-Panis MJ, Teixeira MT, Geli V, Gilson E. CST meets shelterin to keep telomeres in check. Mol Cell 2010; 39:665-76; PMID:20832719; http://dx.doi.org/ 10.1016/j.molcel.2010.08.024 [DOI] [PubMed] [Google Scholar]
- 15. Price CM, Boltz KA, Chaiken MF, Stewart JA, Beilstein MA, Shippen DE. Evolution of CST function in telomere maintenance. Cell Cycle 2010; 9:3157-65; PMID:20697207; http://dx.doi.org/ 10.4161/cc.9.16.12547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen LY, Redon S, Lingner J. The human CST complex is a terminator of telomerase activity. Nature 2012; 488:540-4; PMID:22763445; http://dx.doi.org/ 10.1038/nature11269 [DOI] [PubMed] [Google Scholar]
- 17. Mitton-Fry RM, Anderson EM, Theobald DL, Glustrom LW, Wuttke DS. Structural basis for telomeric single-stranded DNA recognition by yeast Cdc13. J Mol Biol 2004; 338:241-55; PMID:15066429; http://dx.doi.org/ 10.1016/j.jmb.2004.01.063 [DOI] [PubMed] [Google Scholar]
- 18. Taggart AK, Teng SC, Zakian VA. Est1p as a cell cycle-regulated activator of telomere-bound telomerase. Science 2002; 297:1023-6; PMID:12169735; http://dx.doi.org/ 10.1126/science.1074968 [DOI] [PubMed] [Google Scholar]
- 19. Gelinas AD, Paschini M, Reyes FE, Heroux A, Batey RT, Lundblad V, Wuttke DS. Telomere capping proteins are structurally related to RPA with an additional telomere-specific domain. Proc Natl Acad Sci U S A 2009; 106:19298-303; PMID:19884503; http://dx.doi.org/ 10.1073/pnas.0909203106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bryan C, Rice C, Harkisheimer M, Schultz DC, Skordalakes E. Structure of the human telomeric Stn1-Ten1 capping complex. PLoS One 2013; 8:e66756; PMID:23826127; http://dx.doi.org/ 10.1371/journal.pone.0066756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu CC, Gopalakrishnan V, Poon LF, Yan T, Li S. Cdk1 regulates the temporal recruitment of telomerase and cdc13-stn1-ten1 complex for telomere replication. Mol Cell Biol 2014; 34:57-70; PMID:24164896; http://dx.doi.org/ 10.1128/MCB.01235-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li S, Makovets S, Matsuguchi T, Blethrow JD, Shokat KM, Blackburn EH. Cdk1-dependent phosphorylation of Cdc13 coordinates telomere elongation during cell-cycle progression. Cell 2009; 136:50-61; PMID:19135888; http://dx.doi.org/ 10.1016/j.cell.2008.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Puglisi A, Bianchi A, Lemmens L, Damay P, Shore D. Distinct roles for yeast Stn1 in telomere capping and telomerase inhibition. EMBO J 2008; 27:2328-39; PMID:19172739; http://dx.doi.org/ 10.1038/emboj.2008.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qi H, Zakian VA. The Saccharomyces telomere-binding protein Cdc13p interacts with both the catalytic subunit of DNA polymerase alpha and the telomerase-associated est1 protein. Genes Dev 2000; 14:1777-88; PMID:10898792 [PMC free article] [PubMed] [Google Scholar]
- 25. Bianchi A, Shore D. How telomerase reaches its end: mechanism of telomerase regulation by the telomeric complex. Mol Cell 2008; 31:153-65; PMID:18657499; http://dx.doi.org/ 10.1016/j.molcel.2008.06.013 [DOI] [PubMed] [Google Scholar]
- 26. Goulian M, Heard CJ, Grimm SL. Purification and properties of an accessory protein for DNA polymerase alpha/primase. J Biol Chem 1990; 265:13221-30; PMID:2165497 [PubMed] [Google Scholar]
- 27. Casteel DE, Zhuang S, Zeng Y, Perrino FW, Boss GR, Goulian M, Pilz RB. A DNA polymerase-{alpha}{middle dot}primase cofactor with homology to replication protein A-32 regulates DNA replication in mammalian cells. J Biol Chem 2009; 284:5807-18; PMID:19119139; http://dx.doi.org/ 10.1074/jbc.M807593200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Armanios M. An emerging role for the conserved telomere component 1 (CTC1) in human genetic disease. Pediatr Blood Cancer 2012; 59:209-10; PMID:22556055; http://dx.doi.org/ 10.1002/pbc.24200 [DOI] [PubMed] [Google Scholar]
- 29. Walne AJ, Bhagat T, Kirwan M, Gitiaux C, Desguerre I, Leonard N, Nogales E, Vulliamy T, Dokal IS. Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica 2013; 98:334-8; PMID:22899577; http://dx.doi.org/ 10.3324/haematol.2012.071068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Angstadt AY, Thayanithy V, Subramanian S, Modiano JF, Breen M. A genome-wide approach to comparative oncology: high-resolution oligonucleotide aCGH of canine and human osteosarcoma pinpoints shared microaberrations. Cancer Genet 2012; 205:572-87; PMID:23137772; http://dx.doi.org/ 10.1016/j.cancergen.2012.09.005 [DOI] [PubMed] [Google Scholar]
- 31. Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, Haibe-Kains B, Viale G, Delorenzi M, Zhang Y, d'Assignies MS, et al. . Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res 2007; 13:3207-14; PMID:17545524; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-2765 [DOI] [PubMed] [Google Scholar]
- 32. Kao KJ, Chang KM, Hsu HC, Huang AT. Correlation of microarray-based breast cancer molecular subtypes and clinical outcomes: implications for treatment optimization. BMC Cancer 2011; 11:143; PMID:21501481; http://dx.doi.org/ 10.1186/1471-2407-11-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma XJ, Dahiya S, Richardson E, Erlander M, Sgroi DC. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res 2009; 11:R7; PMID:19187537; http://dx.doi.org/ 10.1186/bcr2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. www.Oncomine.Com http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc+GSE2109 Bittner Sarcoma Dataset Summary. [Google Scholar]
- 35. Turashvili G, Bouchal J, Baumforth K, Wei W, Dziechciarkova M, Ehrmann J, Klein J, Fridman E, Skarda J, Srovnal J, et al. . Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer 2007; 7:55; PMID:17389037; http://dx.doi.org/ 10.1186/1471-2407-7-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Calderon-Montano JM, Burgos-Moron E, Orta ML, Lopez-Lazaro M. Effect of DNA repair deficiencies on the cytotoxicity of drugs used in cancer therapy –a review. Curr Med Chem 2014; 21:3419-54; PMID:24934343 [DOI] [PubMed] [Google Scholar]
- 37. Chee CE, Meropol NJ. Current status of gene expression profiling to assist decision making in stage II colon xancer. Oncologist 2014; 19:704-11; PMID:24869929; http://dx.doi.org/ 10.1634/theoncologist.2013-0471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abbotts R, Thompson N, Madhusudan S. DNA repair in cancer: emerging targets for personalized therapy. Cancer Manag Res 2014; 6:77-92; PMID:24600246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst) 2008; 7:686-93; PMID:18243065; http://dx.doi.org/ 10.1016/j.dnarep.2007.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martinez-Marignac VL, Rodrigue A, Davidson D, Couillard M, Al-Moustafa AE, Abramovitz M, Foulkes WD, Masson JY, Aloyz R. The effect of a DNA repair gene on cellular invasiveness: XRCC3 over-expression in breast cancer cells. PLoS One 2011; 6:e16394; PMID:21283680; http://dx.doi.org/ 10.1371/journal.pone.0016394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakanoko T, Saeki H, Morita M, Nakashima Y, Ando K, Oki E, Ohga T, Kakeji Y, Toh Y, Maehara Y. Rad51 expression is a useful predictive factor for the efficacy of neoadjuvant chemoradiotherapy in squamous cell carcinoma of the esophagus. Ann Surg Oncol 2014; 21:597-604; PMID:24065387; http://dx.doi.org/ 10.1245/s10434-013-3220-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jalal S, Earley JN, Turchi JJ. DNA repair: from genome maintenance to biomarker and therapeutic target. Clin Cancer Res 2011; 17:6973-84; PMID:21908578; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gu P, Chang S. Functional characterization of human CTC1 mutations reveals novel mechanisms responsible for the pathogenesis of the telomere disease Coats plus. Aging Cell 2013; 12:1100-9; PMID:23869908; http://dx.doi.org/ 10.1111/acel.12139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol 2009; 11:761-8; PMID:19465921; http://dx.doi.org/ 10.1038/ncb1883 [DOI] [PubMed] [Google Scholar]
- 45. Ying S, Minocherhomji S, Chan KL, Palmai-Pallag T, Chu WK, Wass T, Mankouri HW, Liu Y, Hickson ID. MUS81 promotes common fragile site expression. Nat Cell Biol; 15:1001-7; PMID:23811685; http://dx.doi.org/ 10.1038/ncb2773 [DOI] [PubMed] [Google Scholar]
- 46. Germann SM, Schramke V, Pedersen RT, Gallina I, Eckert-Boulet N, Oestergaard VH, Lisby M. TopBP1/Dpb11 binds DNA anaphase bridges to prevent genome instability. J Cell Biol 2014; 204:45-59; PMID:24379413; http://dx.doi.org/ 10.1083/jcb.201305157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 2009; 138:90-103; PMID:19596237; http://dx.doi.org/ 10.1016/j.cell.2009.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Karnani N, Dutta A. The effect of the intra-S-phase checkpoint on origins of replication in human cells. Genes Dev 2011; 25:621-33; PMID:21406556; http://dx.doi.org/ 10.1101/gad.2029711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ge XQ, Blow JJ. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol 2010; 191:1285-97; PMID:21173116; http://dx.doi.org/ 10.1083/jcb.201007074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010; 37:492-502; PMID:20188668; http://dx.doi.org/ 10.1016/j.molcel.2010.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013; 155:1088-103; PMID:24267891; http://dx.doi.org/ 10.1016/j.cell.2013.10.043 [DOI] [PubMed] [Google Scholar]
- 52. Davies SL, North PS, Hickson ID. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat Struc Mol Biol 2007; 14:677-9; PMID:17603497; http://dx.doi.org/ 10.1038/nsmb1267 [DOI] [PubMed] [Google Scholar]
- 53. Vannier JB, Sandhu S, Petalcorin MI, Wu X, Nabi Z, Ding H, Boulton SJ. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 2013; 342:239-42; PMID:24115439; http://dx.doi.org/ 10.1126/science.1241779 [DOI] [PubMed] [Google Scholar]
- 54. Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem Res Toxicol 2006; 19:1580-94; PMID:17173371; http://dx.doi.org/ 10.1021/tx060164e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Groth P, Auslander S, Majumder MM, Schultz N, Johansson F, Petermann E, Helleday T. Methylated DNA causes a physical block to replication forks independently of damage signalling, O(6)-methylguanine or DNA single-strand breaks and results in DNA damage. J Mol Biol 2010; 402:70-82; PMID:20643142; http://dx.doi.org/ 10.1016/j.jmb.2010.07.010 [DOI] [PubMed] [Google Scholar]
- 56. Hsiang YH, Lihou MG, Liu LF. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res 1989; 49:5077-82; PMID:2548710 [PubMed] [Google Scholar]
- 57. Regairaz M, Zhang YW, Fu H, Agama KK, Tata N, Agrawal S, Aladjem MI, Pommier Y. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J Cell Biol 2011; 195:739-49; PMID:22123861; http://dx.doi.org/ 10.1083/jcb.201104003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Clauson C, Scharer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Biol 2013; 5:a012732; PMID:24086043; http://dx.doi.org/ 10.1101/cshperspect.a012732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang M, Kim JM, Shiotani B, Yang K, Zou L, D'Andrea AD. The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol Cell 2010; 39:259-68; PMID:20670894; http://dx.doi.org/ 10.1016/j.molcel.2010.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tomida J, Itaya A, Shigechi T, Unno J, Uchida E, Ikura M, Masuda Y, Matsuda S, Adachi J, Kobayashi M, et al. . A novel interplay between the Fanconi anemia core complex and ATR-ATRIP kinase during DNA cross-link repair. Nucleic Acids Res 2013; 41:6930-41; PMID:23723247; http://dx.doi.org/ 10.1093/nar/gkt467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem J 2011; 436:527-36; PMID:21615334; http://dx.doi.org/ 10.1042/BJ20102162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sheu YJ, Kinney JB, Lengronne A, Pasero P, Stillman B. Domain within the helicase subunit Mcm4 integrates multiple kinase signals to control DNA replication initiation and fork progression. Proc Natl Acad Sci U S A 2014; 111:E1899-908; PMID:24740181; http://dx.doi.org/ 10.1073/pnas.1404063111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu P, Barkley LR, Day T, Bi X, Slater DM, Alexandrow MG, Nasheuer HP, Vaziri C. The Chk1-mediated S-phase checkpoint targets initiation factor Cdc45 via a Cdc25A/Cdk2-independent mechanism. J Biol Chem 2006; 281:30631-44; PMID:16912045; http://dx.doi.org/ 10.1074/jbc.M602982200 [DOI] [PubMed] [Google Scholar]
- 64. Chen LY, Majerska J, Lingner J. Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev 2013; 27:2099-108; PMID:24115768; http://dx.doi.org/ 10.1101/gad.222893.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Luo YM, Xia NX, Yang L, Li Z, Yang H, Yu HJ, Liu Y, Lei H, Zhou FX, Xie CH, et al. . CTC1 increases the radioresistance of human melanoma cells by inhibiting telomere shortening and apoptosis. Int J Mol Med 2014; 33:1484-90; PMID:24718655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65:55-63; PMID:6606682; http://dx.doi.org/ 10.1016/0022-1759(83)90303-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.