

Abstract
Clusterin (CLU) is known as a multifunctional protein involved in a variety of physiological processes including lipid transport, epithelial cell differentiation, tumorigenesis, and apoptosis. Our recent study has demonstrated that knockdown of clusterin sensitizes pancreatic cancer cell lines to gmcitabine treatment. However the details of this survival mechanism remain undefined. Of the various downstream targets of CLU, we examined activation of the NF-kB transcription factor and subsequent transcriptional regulation of BCL-2 gene in pancreatic cancer cell MIA-PaCa-2. The MIA-PaCa-2 cells were transfected with an antisense oligonucleotide (ASO) against clusterin, which led to a decreased protein level of the antiapoptotic gene BCL-2. Furthermore, inhibition of CLU decreased the function of NF-kB, which is capable of transcriptional regulation of the BCL-2 gene. Inhibiting this pathway increased the apoptotic effect of gmcitabine chemotherapy. Re-activated NF-kB resulted in attenuation of ASO-induced effects, followed by the bcl-2 upregulation, and bcl-2 re-inhibition resulted in attenuation of Re-activated NF-kB -induced effects. Animals injected with ASO CLU in MIA-PaCa-2 cells combined with gmcitabine treatment had fewer tumors than gmcitabine or ASO CLU alone. These findings suggest that knockdown of CLU sensitized MIA-PaCa-2 cells to gmcitabine chemotherapy through modulating NF-Kb/bcl-2 pathway.
Keywords: Pancreatic cancer, chemotherapy, clusterin, NF-kB
Introduction
Pancreatic cancer is a highly lethal malignancy resistant to the apoptosis-inducing effects of radio- and chemotherapy [1]. Currently, gemcitabine appears to be the only clinically active drug but, because of pre-existing or acquired chemoresistance of most of the tumor cells, it failed to significantly improve the outcome of pancreatic carcinoma patients [2]. Identification of the molecular signals that mediate apoptotic resistance in pancreatic cancer is an active area of research.
Clusterin, also known as testosterone-repressed message-2, is overexpressed in a variety of solid tumors [3-9]. The clusterin level also raises in various tissues during cell death responses [10-13]. As clusterin is present during apoptosis, it was initially viewed as a cell death inducer, but other studies suggest that clusterin overproduction occurs in resistant cells [9,14,15]. Thus, clusterin has been described as an anti-apoptotic factor, and it has also been implicated in prostate cancer progression to androgen independence [16]. Miyake et al. [17] have demonstrated that the overexpression of clusterin in human androgen-responsive prostate cancer cells LNCaP by stable transfection rendered them highly resistant to androgen ablation, and the introduction of antisense testosterone-repressed message-2 oligodeoxynucleotide therapy in the Shionogi tumor model induces apoptosis and tumor regression. Moreover inhibition of clusterin gene in pancreatic cancer, osteosarcoma and prostate cancer induces significant reduction of cellular growth and increases cellular apoptosis [9,18,19].
Although clusterin has long been proposed to participate in cell survival and it has been extensively studied to inhibit apoptosis, no studies have been carried out to investigate the link between clusterin and survival signaling pathways. The NF-kB transcription factor appears to be involved in the apoptotic resistance of pancreatic cancer. This is based on observations of constitutive activation of NF-kB in pancreatic cancer cells [20] as well as the fact that inhibition of NF-kB decreases cell survival [21] and enhances the apoptotic effect of chemotherapy in pancreatic cancer cells [22,23]. NF-kB transcription factors can regulate the expression of over 100 different genes dependent on the various functional forms of NF-kB, the stimulus of NF-kB activation and cell type examined. NF-kB has been shown to regulate transcriptionally the expression of several members of the BCL-2 gene family. BCL-2 remains the prototypic antiapoptotic protein and the data are indicated that NF-kB regulates its transcription [24-26]. In pancreatic cancer, BCL-2 is frequently overexpressed and its overexpression confers chemo- and radioresistance and enhances tumorigenic and metastatic capability [27-30]. It remains that NF-kB is involved in the mechanism of BCL-2 transcriptional activation in pancreatic cancer [24]. CLU was once found to inhibit NF-kappaB signaling through stabilization of IkappaBs and that this activity may result in suppression of tumor cell motility [31]. It has recently shown CLU has the activity site involved in NF-kappaB pathway regulation [32]. However, it remains uncertain as to whether CLU is involved in the mechanism of NF-kB/BCL-2 transcriptional activation in pancreatic cancer.
Therefore, given the preliminary cellular studies that demonstrate that the inhibition of CLU sensitises pancreatic cancer to the apoptotic effect of chemotherapy, we sought to determine the molecular events that may mediate this effect. The current study examines the hypothesis that a survival signal from CLU activation is mediated by NF-kB and subsequent transcriptional regulation of BCL-2 gene; furthermore, inhibition of this pathway sensitises pancreatic cancer cells to the apoptotic effect of gemcitabine.
Materials and methods
Antibodies
The following primary and secondary antibodies were purchased from Santa Cruz Biotechnology (CA, USA): Anti-NF-kB p65, Anti-Bcl-2 and Anti-CLU. The following fluorescent secondary antibodies were purchased from Molecular Probes (Eugene, Oregon, USA): Alexa fluor 488 goat anti-rabbit IgG, Alexa Fluor 488 rabbit anti-goat IgG and Texas Red-X goat anti-mouse IgG.
Cell culture
The human pancreatic adenocarcinoma cell lines MIA-PaCa-2 were obtained from the American Type Culture Collection (Rockville, MD, USA). The MIA-PaCa-2 cell line has been previously demonstrated to be resistant to gemcitabine [24]. Cells were routinely cultured in DMEM supplemented with 10% fetal bovine serum in a 37°C incubator in a humidified atmosphere of 5% CO2. The cells underwent serum starvation for the 24 h prior to treatment with gmcitabine or transient transfection.
Antisense oligonucleotides and transfection
Second-generation 2’ methoxyethyl gapmer oligonucleotides used in this study were supplied by Dr. CHEN [9]. The sequence of ASO that corresponds to human clusterin translation initiation site was 5’-CAGCAGCAGAGTCTTCATCAT-3’. The mismatch control oligo was 5’-CAGCAGCAGAGTATTTATCAT-3’. Subconflunet MIA-PaCa-2 cells were transfected with either oligo by use of a mixture of Lipofectin (Gibco, Gaithersburg, MD) or oligos in Opti-MEM media (Gibco) at a ratio of 3 µL Lipofectin/mL media per 100 nM oligo. After 5 hours of incubation, cells were replaced by the regular complete media.
Flow cytometry
7-Aminoactinomycin D (7-AAD) is a fluorescent DNA-binding stain that is used to detect apoptotic cells by flow cytometry. Percent apoptosis was measured by use of 7-AAD (Molecular Probes, Eugene, OR) with flow cytometry. Cells (2×105) were plated into 25-cm2 flasks for each data point. After 24 hours of 37°C incubation, the cells were classified into several groups: ① transfected with 500 nM ASO and missense (MS) oligonucleotide; ② treated with 50 ng/ml TNF-α (Roche Applied Science) and 500 nM ASO or missense (MS) oligonucleotide; ③ transfected with 500 nM ASO or missense (MS) oligonucleotide and 50 ng/ml TNF-α and 40 µM TW-37 (a negative regulator of Bcl-2). Cells above were then treated with 0.05-20 µmol/ml gmcitabine the following day. At 72 hours after gmcitabine, cells were harvested and stained with 7-AAD. The stained cells were fixed with 1% paraformaldehyde and analyzed by FACScan. Cells with intermediate levels of 7-AAD staining were scored as apoptotic.
MTT assay
Briefly, cells were seeded at a density of 2,000 to 5,000 cells/well in 96 well plates and grown overnight. They were subject to various treatments. 72 hours later, 20 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/mL in PBS) were added to each well and incubated further for 2 hours. Upon termination, the supernatant was aspirated and the MTT formazan formed by metabolically viable cells was dissolved in 100 µL of isopropanol. The plates were mixed for 30 minutes on a gyratory shaker, and absorbance was measured at 595 nm using a plate reader.All samples were done in triplicate, and data were analyzed by Student’s t test.
Preparation of nuclear and cytoplasmic extracts
Nuclear and cytoplasmic soluble extracts were prepared from the cells in various groups in various time point using a rapid version of the method as previously described [33,34]. Cytoplasmic extracts were obtained by diluting the supernatant obtained after the first centrifugation with 3 vol. of buffer D [34].
Detection of NF-KB binding activity by electrophoretic mobility shift assay (EMSA)
Nuclear protein extracts were prepared as described previously [33,34]. The sequence of the NF-kB oligonucleotide probe was 5’-AGTTGAGGGACTTTCCCAGGC-3’. EMSA was performed as described previously [33,34].
Western blot assay
Total cellular proteins were isolated and the protein concentration of the sample was determined by BioRad DC Protein Assay (Bio-Rad Laboratories Inc., Hercules, CA). CLU, NF-kB p65, bcl-2 and β-actin were detected. The targeted protein was revealed by enhanced chemiluminescence (ECL). The membrane was incubated with an ECL solution (Biological Industries) and exposed to ECL film (Eastman Kodak, Rochester, NY) to visualize specifically labeled proteins. The resulting exposed films were then analyzed by densitometry. All experiments were performed at least three times.
In vivo experimental growth assays
Female mice at 4-6 weeks old were obtained from Tianjin Medical University Cancer Institute for tumor implantation. All animals were maintained in a sterile environment and cared for within the laboratory animal regulations of the Ministry of Science and Technology of the People’s Republic of China (http://www.most.gov.cn/kytj/kytjzcwj/200411). Full details of the study approval by the ethics committee at the Tianjin Medical university Cancer Institute and Hospital.A suspension of 2×106 cells in 50 µL volume was injected subcutaneously into the left posterior flank of mice by use of a 1-cc syringe with 27½-gauge needle. Tumors were grown for 14-21 days until average tumor volume reached 0.1 cm3. Treatment groups consisted of untreated control (PBS), ASO mock treatment alone, ASO mock plus gmcitabine treatment, ASO CLU alone, and ASO CLU plus gmcitabine treatment. Each treatment group contained 7 mice. ASO mock and ASO CLU at doses of 10 mg/kg were administered by iv injection, and every other day for a total of seven injections, whereas gemcitabine dose was based on previously published reports (80 mg/kg body weight, i.v., and every other day for a total of three injections). The experiments were terminated 28 days Tumor growth was monitored thrice a week by calipers to calculate tumor volumes according to the formula [length × width2]/2. Tumor weights were determined at the end of the study when mice were sacrificed. All experiments were repeated at least twice. Representative experiments are shown.
In vivo apoptosis assay using TdT-mediated nick end labelling (TUNEL)
Apoptosis was measured using a commercially available ApopTag Apoptosis Detection Kit as per the instructions of the manufacturer. Staining intensity and localization for terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling was scored by two investigators independently.
Statistical assessment
All statistical analyses were performed using SPSS13.0 software. The results were presented as mean ± SD of three replicate assays. Differences between various groups were assessed using ANOVA or Dunnett t-test. A P value of <0.05 was considered to indicate statistical significance.
Results
In vitro targeted knockdown of CLU alone did not promote cell apoptosis and inhibit proliferation
Western blotting revealed that the MIA-PaCa-2 cell line showed a higher basal level protein expression of CLU, P65 and bcl-2, evaluated by western blot analysis (Figure 1A). MIA-PaCa-2 cells treated with various concentrations of ASO CLU (50, 500, or 1000 n m) for 48 h significantly reduced CLU, P65 and bcl-2 expression levels (Figure 1A), and NF-Kb activity (Figure 1B). In contrast, CLU, P65 and bcl-2 expression levels were not affected by ASO control at any of the used concentrations (data not shown). We then examined the effects of ASO CLU or ASO control treatment on apoptosis and growth in the MIA-PaCa-2 cells. As shown in Figure 1C and 1D, ASO CLU or ASO control treatment did not significantly induce apoptosis and inhibit growth in MIA-PaCa-2 cells (P>0.05, respectively).
Figure 1.
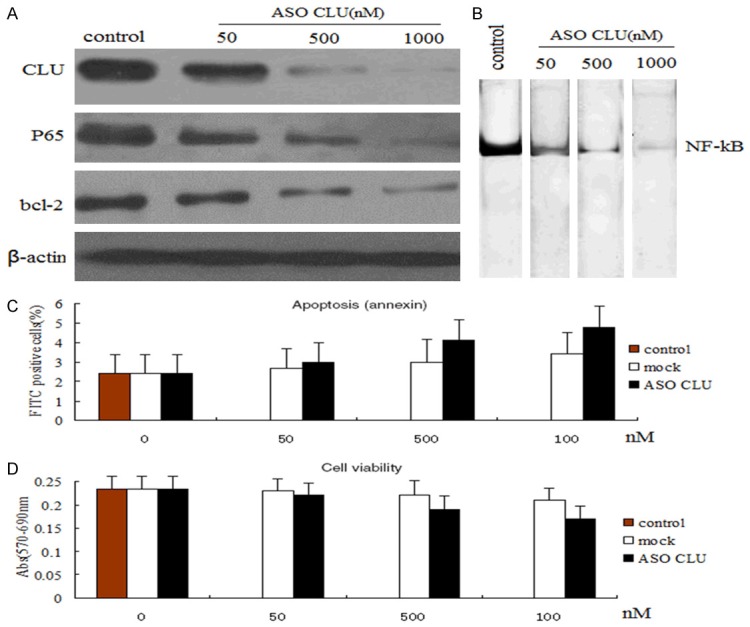
Effect of ASO CLU on cell viability and apoptosis of MIA-PaCa-2 cells. A. Western blot analysis of CLU, NF-kB and bcl-2 in whole cell lysates or nuclear of MIA-PaCa-2 cells after treatment with various concentrations (50-1000 nM) of ASO CLU for 48 h. Down-regulation of CLU, NF-kB and bcl-2 expression levels is ASO CLU dose-dependant. β-actin protein as loading control is shown for each blot. 1000 nM ASO CLU could almost completely inhibit the CLU, NF-kB and bcl-2 expression. B. NF-kB DNA binding activity in the nuclear extract of the investigated MIA-PaCa-2 cells in the presence of various concentrations (50-1000 nM) of ASO CLU for 48 h. ASO CLU down-regulated the NF-Kb activity in MIA-PaCa-2 cells. C. Knockdown of CLU failed to induce any significant changes in cell apoptisis of MIA-PaCa-2 cells. D. Knockdown of CLU failed to induce any significant changes in cell viability of MIA-PaCa-2 cells.
In vitro gmcitabine treatment did not activated CLU and NF-kB activity
It has reported MIAPaCa-2 cells were resistant to gemcitabine treatment [24], and CLU confers gemcitabine resistance in pancreatic cancer [9]. Therefore, we wished to evaluate whether CLU could be activited in response to treatment with gemcitabine.MIAPaCa-2 cells were stimulated with various concentrations (0.05, 5, 20 µmol/L) of gemcitabine for 24 hrs in vitro and the expression of CLU, P65 and bcl-2 was evaluated by western blot analysis and NF-kB activities were measured using EMSA assay. Gemcitabine treatment (0.05, 5 and 20 µ umol/L) for 24 hours had no significant effect on CLU, P65 and bcl-2 (Figure 2A) and NF-kB activities (Figure 2B).
Figure 2.
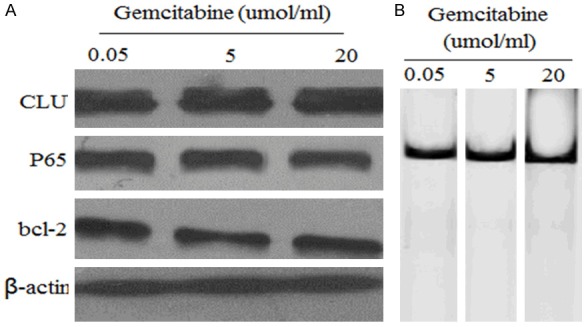
Effect of gemcitabine on CLU, NF-kB and bcl-2 protein and NF-kB activity. (A) Western blot analysis of CLU, NF-kB and bcl-2 in whole cell lysates or nuclear of MIA-PaCa-2 cells after treatment with various concentrations (0.05-20 µmol/ml) of gemcitabine for 24 h. (B) EMSA analysis of NF-kB activity in MIA-PaCa-2 cells after treatment with various concentrations (0.05-20 µmol/ml) of gemcitabine for 24 h. No significant effect on CLU, P65 and bcl-2 (A) and NF-kB activities was found after treatment with various concentrations (0.05-20 µmol/ml) of gemcitabine.
In vitro knockdown of CLU influenced sensitive to gemcitabine
We next assessed the effect of a combination of ASO CLU and gemcitabine on cell viability and apoptosis by MTT and ELISA assay. For these studies, MIAPaCa-2 cells were pretreated with 500 nM ASO CLU or ASO control for 24 h, then the cells were stimulated with 20 µmol/L of gemcitabine for 72 hrs in vitro, and viable cells and apoptosic cells were evaluated at 96 hours posttreatment by MTT assay or ELISA assay. The dose used here was chosen based upon a preliminary dose escalation study done by us. We found that treatment with ASO CLU in combination with gemcitabine for 72 hours resulted in 75% loss of viability of MIAPaCa-2 cells (Figure 3A), and 41% increase in apoptosic cells (Figure 3B). However, treatment with ASO control plus gemcitabine simultaneously did not have significant effect compared to gemcitabine alone (Figure 3A and 3B).
Figure 3.
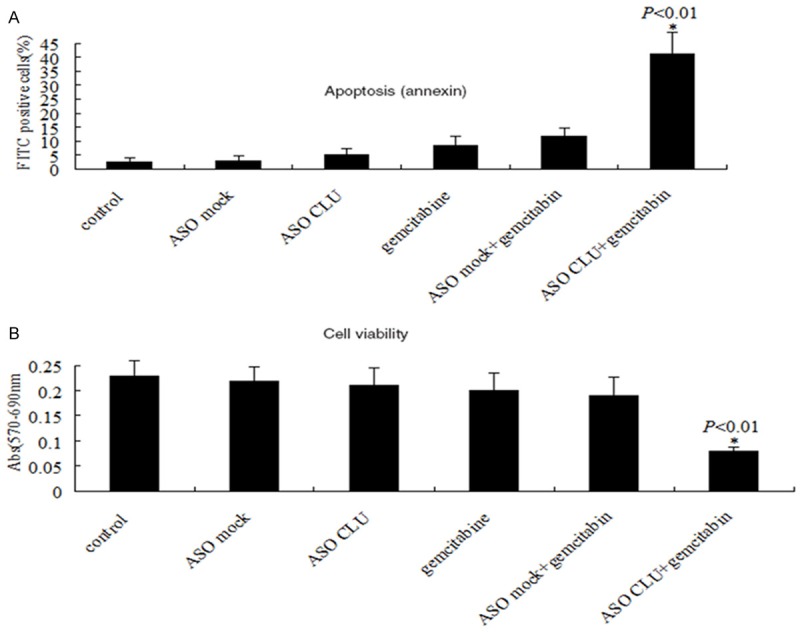
ASO CLU influenced sensitive to gemcitabine in MIAPaCa-2 cells. A. ELISA assay: Treatment of MIAPaCa-2 cells with 500 nM ASO CLU or ASO control in combination with gemcitabine for 72 hours induced a significant increase in apoptosic cells. B. MTT assay: Treatment of MIAPaCa-2 cells with 500 nM ASO CLU or ASO control in combination with gemcitabine for 72 hours induced a significant decrease in viability. *P<0.01 vs. every other groups.
In vitro knockdown of CLU influenced sensitive to gemcitabine via NF-kB/bcl-2 dependant pathway
We have observed a decrease in P65 and bcl-2 protein levels (Figure 2A), and P65 activity (Figure 2B) in MIAPaCa-2 cells when CLU was knockdown by ASO CLU, followed by the increase of sensitity to gemcitabine. Next, we analyzed whether re-activation of NF-KB by TNF-α could restore the chemoresistant phenotype of MIAPaCa-2 cells above. MIAPaCa-2 cells were transfected with 500 nM ASO or ASO control for 24 h, then treated with 50 ng/ml TNF-α for another 16 h, later, cells above were then treated with 20 µmol/ml gmcitabine for 72 hrs. Our results above showed that NF-kB activity inhibited by ASO CLU could be promoted by TNF-α treatment, and combination of TNF-α treatment induced much less apoptosis in MIAPaCa-2 cells relative to knockdown of CLU pretreatment followed by gemcitabine treatment as shown by ELISA (Figure 4B). Promotion of cell growth and viability as assessed by MTT could also be due to the reduction of apoptotic cell death induced by TNF-α treatment (Figure 4A). The above results clearly suggest that the enhanced cell growth inhibition and induction of apoptosis by CLU sliencing in combination with gmcitabine in MIAPaCa-2 cells is NF-kB-dependant. Previous study found Bcl-2 remains the prototypic antiapoptotic protein and NF-kB regulates its transcription. Next, we analyzed whether NF-kB-dependant Bcl-2 regulation is required for chemosensitisation in MIAPaCa-2 cells. Our results shown knockdown of CLU inhibited the p65 expression and activity (Figure 4D), followed by the bcl-2 inhibition (Figure 4C). TNF-α treatment enhanced the p65 expression (Figure 4C) and activity (Figure 4D), followed by the bcl-2 upregulation (Figure 4C). However, when MIAPaCa-2 cells (transfected with 500 nM ASO + 50 ng/ml TNF-α + 20 µmol/ml + gmcitabine) were treated with 40 µM TW-37 (a negative regulator of Bcl-2) for 12 hs to inhibit Bcl-2 expression, cell growth inhibition (Figure 4B) and induction of apoptosis (Figure 4A) was significantly enhanced. These results show that CLU down-regulates NF-kB DNA-binding activity and NF-kB-induced bcl-2 expression, which could be the molecular mechanism of gemcitabine-induced cell death in CLU sliencing-pretreated cells.
Figure 4.

ASO CLU influenced sensitive to gemcitabine via NF-kB/bcl-2 dependant pathway. A. ELISA analysis of apoptosis in MIAPaCa-2 cells after treatment with ASO CLU + gemcitabine, ASO CLU + gemcitabine + TNF-α and ASO CLU + gemcitabine + TNF-α + TW-37. Restoration of the NF-kB activity by TNF-α treatment induced a significant decrease in apoptosic cells, and re-inhibition of the bcl-2 expression by TW-37 inTNF-α treated MIAPaCa-2 cells induced a significant increase in apoptosic cells. B. MTT analysis of viable cells, which has the similar results as above. C. Western blot analysis of CLU, NF-kB and bcl-2 in whole cell lysates or nuclear of MIA-PaCa-2 cells above. D. EMSA analysis of NF-kB DNA binding activity in the nuclear extract of the investigated MIA-PaCa-2 cells above.
ASO CLU augments in vivo therapeutic effect of gemcitabine on primary tumor
The experiment was done to investigate the therapeutic utility of ASO CLU and gemcitabine combination in SCID mice bearing subcutaneously implanted pancreatic tumor cells MIAPaCa-2. ASO mock and ASO CLU at doses of 10 mg/kg were administered by iv injection, and every other day for a total of seven injections, whereas gemcitabine dose was based on previously published reports (80 mg/kg body weight, i.v., and every other day for a total of three injections). For in vivo experiment, 42 mice were divided into six groups as described in Materials and Methods. In an effort to establish the efficacy of a single-agent treatment compared with combinations, we determined the mean tumor weight in all treated groups. Analysis of the average tumor weight in the different treatment groups showed that single modality treatment with either ASO CLU or gemcitabine alone in mice harboring MIAPaCa-2 cells caused 12% and 16% reduction in tumor weight, respectively, compared with control tumors (Figure 5A). Under identical experimental conditions, combination of ASO CLU and gemcitabine treatment showed significant decrease (70%, P<0.01) in tumor weight compared with untreated control, ASO CLU alone, ASO mock alone, gemcitabine alone or ASO mock in combination with gemcitabine treated group. Furthermore, combination of ASO CLU and gemcitabine treatment resulted in a significantly decrease in the MIAPaCa-2 tumor volume (Figure 5B). Figure 5C, 5D shows that combination of ASO CLU and gemcitabine treatment induced significantly tumor cell apoptosis as measured by TUNEL staining.
Figure 5.
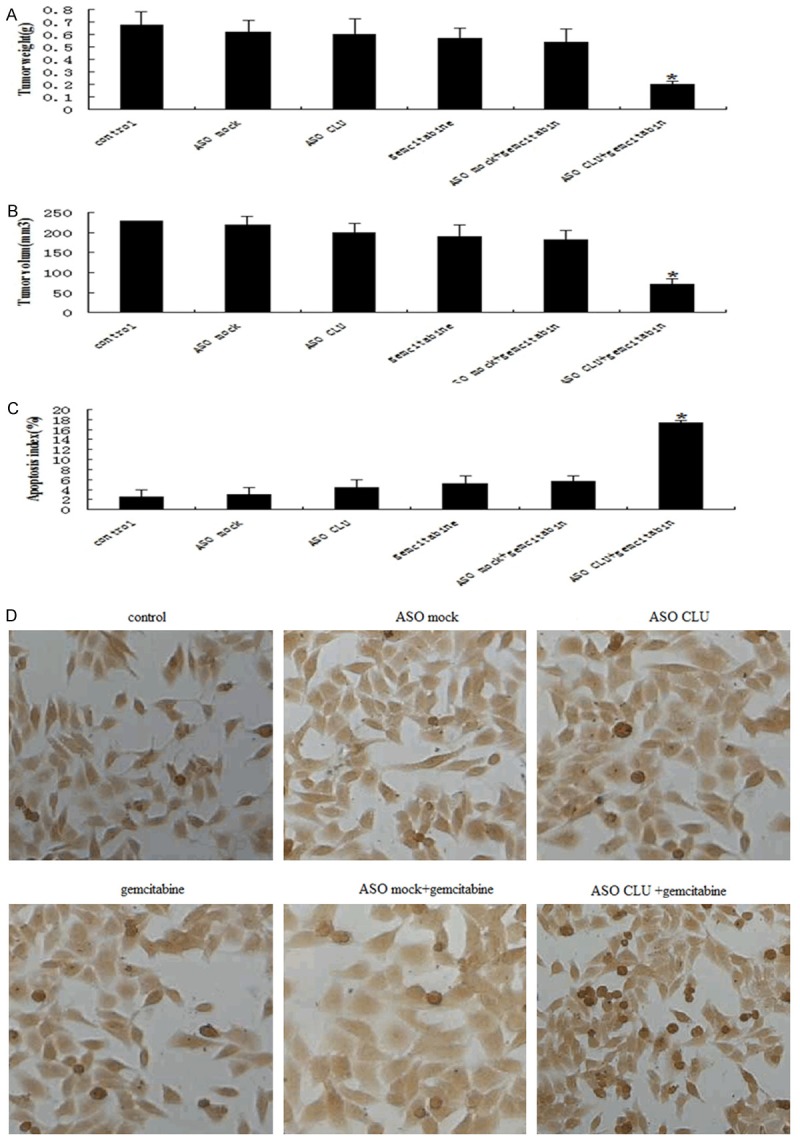
Effect of ASO CLU combined with gemcitabine on primary tumor. Xenograft tumors were established by s.c. injection of MIAPaCa-2 2×106 (2×106) in the flank of each nude mouse. When the tumor was ~50-100 mm3, treatment groups consisted of blank control groups, ASO mock control groups, ASO mock + gmcitabine groups, ASO CLU groups, and ASO CLU + gmcitabine groups. ASO mock and ASO CLU at doses of 10 mg/kg were administered by iv injection, and every other day for a total of seven injections, whereas gemcitabine dose was based on previously published reports (80 mg/kg body weight, i.v., and every other day for a total of three injections). The experiments were terminated 28 days. Tumor weights were determined at the end of the study when mice were sacrificed. All experiments were repeated at least twice. Representative experiments are shown. Tumor volume was monitored thrice a week by calipers to calculate tumor volumes according to the formula [length × width2]/2. A. Representative histogram showing the tumor weight in different groups. B. Representative histogram showing the tumor volume in different groups. C. Apoptosic index in different groups. D. Representative histogram showing the TUNEL positive cells in different groups. Each bar represents mean + SE; *P<0.01. All experiments were repeated three times with similar results.
Discussion
Inhibition of the CLU has repeatedly and consistently been shown to sensitize pancreatic cancer cells in vitro and in vivo to the apoptotic effect of chemotherapy [9]. The mechanism by which CLU activation in these cancer cells confers chemoresistance is unclear. CLU has been shown to regulate multiple targets of apoptosis, including Bax [35,36], Bcl-XL [37] and p21 [38]. There has been extensive investigation coupling CLU to Ikk and NF-kB activation [31,32], which can be a potent survival signal through transcription of various target genes. However, which genes confer this effect is undergoing further investigation. The effect of modifying single members of the BCL-2 gene family on apoptotic response has been examined in a variety of cancer types and models, and NF-kB has been shown to regulate transcriptionally the expression of several members of the BCL-2 gene family [24-26].
While we previously demonstrated that expression of CLU conferred a chemoresistant in pancreatic cancer cells [9], a direct correlation between CLU and chemoresistant has not been demonstrated. Thus, it is of interest to determine how CLU contributes to the gemcitabine chemoresistant of pancreatic cancer cells.
In our study, knockdown of CLU could attenuate gmcitabine-induced apoptosis and growth inhibition in vivo and vitro, suggesting that knockdown of endogenous CLU is a key determinant of gmcitabine-induced apoptosis. A recent study has shown that blockage of endogenous CLU expression reduced viability and enhanced apoptosis in AR4-2J cells. Presence of CLU reduced NF-kappaB activation and nuclear translocation and expression of the NF-kappaB target genes TNF-alpha and MOB-1 under cell stress [39]. Our current study provides evidence that knockdown of CLU itself could not promote apoptosis and inhibit growth in MIA-PaCa-2 cells, however knockdown of CLU is required for the sensitivity of pancreatic cancer cells to gmcitabine-induced apoptosis. Although expression of CLU and NF-kB conferred a chemoresistant in pancreatic cancer cells, gmcitabine treatment did not induce CLU and NF-kB upregulation. One of the most remarkable findings is that the NF-kB was mediated by CLU. Furthermore, reintroduction of NF-kB activity and nuclear translocation in the presence of TNF-α restored the chemoresistant in pancreatic cancer cells, though CLU was inhibited, suggesting that knockdown of endogenous CLU sensitized pancreatic cancer cells to gmcitabine via NF-kB-dependant pathway.
Other remarkable findings are that the bcl-2-anti-apoptosic effect by CLU was mediated by NF-kB. Furthermore, reinhibition of bcl-2 by TW-37 (a negative regulator of Bcl-2) deteriorated the chemoresistance of reintroduction of NF-kB activity in pancreatic cancer cell, suggesting that bcl-2 is a key determinant of gmcitabine-induced apoptosis. These data support the hypothesis that CLU regulates chemosensitivity in pancreatic cancer via regulating NF-KB/bcl-2 pathway. These data may provide a functional link between elevated CLU levels observed in human pancreatic cancer patients and poor prognosis [40].
In summary, we have demonstrated that knockdown of CLU is essential for gmcitabine-induced apoptosis in human pancreatic cancer cell, and that this is mediated, at least in part, through the downregulation of NF-Kb and bcl-2. Activation of CLU confers resistance by promoting NF-kB-mediated transactivation and bcl-2 upregulation.
A more thorough understanding of the molecular mechanisms underling chemosensitivity in human pancreatic cancer may ultimately improve treatment outcomes for this disease.
Acknowledgements
This work was supported by the natural science research fund from Shandong province (No. 2014ZRB01298). This study was conducted in the central Lab of Weifang Medical College and Clinical Lab of people’s hospital of Weifang.
Disclosure of conflict of interest
None.
References
- 1.Squadroni M, Fazio N. Chemotherapy in pancreatic adenocarcinoma. Eur Rev Med Pharmacol Sci. 2010;14:386–94. [PubMed] [Google Scholar]
- 2.Arlt A, Gehrz A, Müerköster S, Vorndamm J, Kruse ML, Fölsch UR, Schäfer H. Role of NFkappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene. 2003;22:3243–51. doi: 10.1038/sj.onc.1206390. [DOI] [PubMed] [Google Scholar]
- 3.Redondo M, Rodrigo I, Alcaide J, Tellez T, Roldan MJ, Funez R, Diaz-Martin A, Rueda A, Jiménez E. Clusterin expression is associated with decreased disease-free survival of patients with colorectal carcinomas. Histopathology. 2010;56:932–6. doi: 10.1111/j.1365-2559.2010.03565.x. [DOI] [PubMed] [Google Scholar]
- 4.Bi J, Guo AL, Lai YR, Li B, Zhong JM, Wu HQ, Xie Z, He YL, Lv ZL, Lau SH, Wang Q, Huang XH, Zhang LJ, Wen JM, Guan XY. Overexpression of clusterin correlates with tumor progression, metastasis in gastric cancer: a study on tissue microarrays. Neoplasma. 2010;57:191–7. doi: 10.4149/neo_2010_03_191. [DOI] [PubMed] [Google Scholar]
- 5.Flanagan L, Whyte L, Chatterjee N, Tenniswood M. Effects of clusterin over-expression on metastatic progression and therapy in breast cancer. BMC Cancer. 2010;10:107. doi: 10.1186/1471-2407-10-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang GF, Li XM, Xie D. Overexpression of clusterin in ovarian cancer is correlated with impaired survival. Int J Gynecol Cancer. 2009;19:1342–6. doi: 10.1111/IGC.0b013e3181a83ed9. [DOI] [PubMed] [Google Scholar]
- 7.Albert JM, Gonzalez A, Massion PP, Chen H, Olson SJ, Shyr Y, Diaz R, Lambright ES, Sandler A, Carbone DP, Putnam JB Jr, Johnson DH, Lu B. Cytoplasmic clusterin expression is associated with longer survival in patients with resected non small cell lung cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:1845–51. doi: 10.1158/1055-9965.EPI-07-0146. [DOI] [PubMed] [Google Scholar]
- 8.Kantoff P. Recent progress in management of advanced prostate cancer. Oncology. 2005;19:631–6. [PubMed] [Google Scholar]
- 9.Chen Q, Wang Z, Zhang K, Liu X, Cao W, Zhang L, Zhang S, Yan B, Wang Y, Xia C. Clusterin confers gmcitabine resistance in pancreatic cancer. World J Surg Oncol. 2011;9:59. doi: 10.1186/1477-7819-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bursch W, Gleeson T, Kleine L, Tenniswood M. Expression of clusterin (testosterone-repressed prostate message-2) mRNA during growth and regeneration of rat liver. Arch Toxicol. 1995;69:253–8. doi: 10.1007/s002040050167. [DOI] [PubMed] [Google Scholar]
- 11.Youm YH, Yang H, Yoon YD, Kim DY, Lee C, Yoo TK. Doxazosin-induced clusterin expression and apoptosis in prostate cancer cells. Urol Oncol. 2007;25:483–8. doi: 10.1016/j.urolonc.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 12.Kyprianou N, English HF, Davidson NE, Isaacs JT. Programmed cell death during regression of the MCF-7 human breast cancer following estrogen ablation. Cancer Res. 1991;51:162–6. [PubMed] [Google Scholar]
- 13.Kyprianou N, English HF, Isaacs JT. Programmed cell death during regression of PC-82 human prostate cancer following androgen ablation. Cancer Res. 1990;50:3748–53. [PubMed] [Google Scholar]
- 14.French LE, Sappino AP, Tschopp J, Schifferli JA. Distinct sites of production and deposition of the putative cell death marker clusterin in the human thymus. J Clin Invest. 1992;90:1919–25. doi: 10.1172/JCI116069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho SM, Leav I, Ghatak S, Merk F, Jagannathan VS, Mallery K. Lack of association between enhanced TRPM-2/clusterin expression and increased apoptotic activity in sex-hormone-induced prostatic dysplasia of the Noble rat. Am J Pathol. 1998;153:131–9. doi: 10.1016/S0002-9440(10)65553-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.July LV, Akbari M, Zellweger T, Jones EC, Goldenberg SL, Gleave ME. Clusterin expression is significantly enhanced in prostate cancer cells following androgen withdrawal therapy. Prostate. 2002;50:179–88. doi: 10.1002/pros.10047. [DOI] [PubMed] [Google Scholar]
- 17.Miyake H, Nelson C, Rennie PS, Gleave ME. Testosterone-repressed prostate message-2 is an antiapoptotic gene involved in progression to androgen independence in prostate cancer. Cancer Res. 2000;60:170–6. [PubMed] [Google Scholar]
- 18.Miyake H, Nelson C, Rennie PS, Gleave ME. Acquisition of chemoresistant phenotype by overexpression of the antiapoptotic gene testosterone-repressed prostate message-2 in prostate cancer xenograft models. Cancer Res. 2000;60:2547–54. [PubMed] [Google Scholar]
- 19.Trougakos IP, So A, Jansen B, Gleave ME, Gonos ES. Silencing expression of the clusterin/apolipoprotein j gene in human cancer cells using small interfering RNA induces spontaneous apoptosis, reduced growth ability, and cell sensitization to genotoxic and oxidative stress. Cancer Res. 2004;64:1834–42. doi: 10.1158/0008-5472.can-03-2664. [DOI] [PubMed] [Google Scholar]
- 20.Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–127. [PubMed] [Google Scholar]
- 21.Shah SA, Potter MW, McDade TP, Ricciardi R, Perugini RA, Elliott PJ, Adams J, Callery MP. 26S proteasome inhibition induces apoptosis and limits growth of human pancreatic cancer. J Cell Biochem. 2002;82:110–122. doi: 10.1002/jcb.1150. [DOI] [PubMed] [Google Scholar]
- 22.Arlt A, Vorndamm J, Breitenbroich M, Folsch UR, Kalthoff H, Schmidt WE, Schafer H. Inhibition of NF-kappaB sensitizes human pancreatic carcinoma cells to apoptosis induced by etoposide (VP16) or doxorubicin. Oncogene. 2001;20:859–868. doi: 10.1038/sj.onc.1204168. [DOI] [PubMed] [Google Scholar]
- 23.Kong R, Sun B, Jiang H, Pan S, Chen H, Wang S, Krissansen GW, Sun X. Downregulation of nuclear factor-kappaB p65 subunit by small interfering RNA synergizes with gemcitabine to inhibit the growth of pancreatic cancer. Cancer Lett. 2010;291:90–8. doi: 10.1016/j.canlet.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Fahy BN, Schlieman M, Virudachalam S, Bold RJ. AKT inhibition is associated with chemosensitisation in the pancreatic cancer cell line MIA-PaCa-2. Br J Cancer. 2003;89:391–397. doi: 10.1038/sj.bjc.6601037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao X, Ning Q, Sun X, Tian D. Pokemon reduces Bcl-2 expression through NF-κ Bp65: A possible mechanism of hepatocellular carcinoma. Asian Pac J Trop Med. 2011;4:492–497. doi: 10.1016/S1995-7645(11)60133-8. [DOI] [PubMed] [Google Scholar]
- 26.Chu SH, Lim JW, Kim DG, Lee ES, Kim KH, Kim H. Down-regulation of Bcl-2 is mediated by NF-κB activation in Helicobacter pylori-induced apoptosis of gastric epithelial cells. Scand J Gastroenterol. 2011;46:148–55. doi: 10.3109/00365521.2010.525255. [DOI] [PubMed] [Google Scholar]
- 27.Bold RJ, Chandra J, McConkey DJ. Gemcitabine-induced programmed cell death (apoptosis) of human pancreatic carcinoma is determined by Bcl-2 content. Ann Surg Oncol. 1999;6:279–285. doi: 10.1007/s10434-999-0279-x. [DOI] [PubMed] [Google Scholar]
- 28.Bold RJ, Hess KR, Pearson AS, Grau AM, Sinicrope FA, Jennings M, McConkey DJ, Bucana CD, Cleary KR, Hallin PA, Chiao PJ, Abbruzzese JL, Evans DB. Prognostic factors in resectable pancreatic cancer: p53 and bcl-2. J Gastrointest Surg. 1999;3:263–277. doi: 10.1016/s1091-255x(99)80068-7. [DOI] [PubMed] [Google Scholar]
- 29.Nio Y, Dong M, Iguchi C, Yamasawa K, Toga T, Itakura M, Tamura K. Apoptosis and expression of Bcl-2 and Bax proteins in invasive ductal carcinoma of the pancreas. J Surg Oncol. 2001;76:188–196. doi: 10.1002/jso.1033. [DOI] [PubMed] [Google Scholar]
- 30.Su Z, Lebedeva IV, Gopalkrishnan RV, Goldstein NI, Stein CA, Reed JC, Dent P, Fisher PB. A combinatorial approach for selectively inducing programmed cell death in human pancreatic cancer cells. Proc Natl Acad Sci U S A. 2001;98:10332–10337. doi: 10.1073/pnas.171315198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santilli G, Aronow BJ, Sala A. Essential requirement of apolipoprotein J (clusterin) signaling for IkappaB expression and regulation of NF-kappaB activity. J Biol Chem. 2003;278:38214–9. doi: 10.1074/jbc.C300252200. [DOI] [PubMed] [Google Scholar]
- 32.Essabbani A, Margottin-Goguet F, Chiocchia G. Identification of clusterin domain involved in NF-kappaB pathway regulation. J Biol Chem. 2010;285:4273–7. doi: 10.1074/jbc.C109.057133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci U S A. 1989;86:2336–2340. doi: 10.1073/pnas.86.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trougakos IP, Lourda M, Antonelou MH, Kletsas D, Gorgoulis VG, Papassideri IS, Zou Y, Margaritis LH, Boothman DA, Gonos ES. Intracellular clusterin inhibits mitochondrial apoptosis by suppressing p53-activating stress signals and stabilizing the cytosolic Ku70-Bax protein complex. Clin Cancer Res. 2009;15:48–59. doi: 10.1158/1078-0432.CCR-08-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Kim JK, Edwards CA, Xu Z, Taichman R, Wang CY. Clusterin inhibits apoptosis by interacting with activated Bax. Nat Cell Biol. 2005;7:909–15. doi: 10.1038/ncb1291. [DOI] [PubMed] [Google Scholar]
- 37.Kim N, Yoo JC, Han JY, Hwang EM, Kim YS, Jeong EY, Sun CH, Yi GS, Roh GS, Kim HJ, Kang SS, Cho GJ, Park JY, Choi WS. Human nuclear clusterin mediates apoptosis by interacting with Bcl-XL through C-terminal coiled coil domain. J Cell Physiol. 2011;227:1157–67. doi: 10.1002/jcp.22836. [DOI] [PubMed] [Google Scholar]
- 38.Chen T, Turner J, McCarthy S, Scaltriti M, Bettuzzi S, Yeatman TJ. Clusterin-mediated apoptosis is regulated by adenomatous polyposis coli and is p21 dependent but p53 independent. Cancer Res. 2004;64:7412–9. doi: 10.1158/0008-5472.CAN-04-2077. [DOI] [PubMed] [Google Scholar]
- 39.Savković V, Gantzer H, Reiser U, Selig L, Gaiser S, Sack U, Klöppel G, Mössner J, Keim V, Horn F, Bödeker H. Clusterin is protective in pancreatitis through anti-apoptotic and anti-inflammatory properties. Biochem Biophys Res Commun. 2007;356:431–7. doi: 10.1016/j.bbrc.2007.02.148. [DOI] [PubMed] [Google Scholar]
- 40.Xie MJ, Motoo Y, Su SB, Mouri H, Ohtsubo K, Matsubara F, Sawabu N. Expression of clusterin in human pancreatic cancer. Pancreas. 2002;25:234–8. doi: 10.1097/00006676-200210000-00004. [DOI] [PubMed] [Google Scholar]