

Abstract
Smoking is a well-known risk factor for many systemic diseases and oral disorders. Smoking has been recognized to cause diminished defense, persistent inflammation and result in disease development. Nucleotide binding oligomerization domain 1 (NOD1) signal pathway plays a key role in innate immune and tissue homeostasis. Our recent studies confirmed that cigarette smoke extract (CSE) could inhibit NOD1 expression and affect expression levels of crucial molecules of NOD1 signaling in oral mucosal epithelial cells. In the present study, immortalized human oral mucosal epithelial (Leuk-1) cells were treated with CSE, iE-DAP (NOD1 agonist), CSE + iE-DAP, respectively. Western blotting analysis demonstrated that iE-DAP triggered NOD1 expression of leuk-1 cells in a dose-dependent manner. iE-DAP also reversed the suppressive effect of CSE on NOD1 expression and prevented the overactivation of RIP2 and P-NF-κB following CSE exposure. Real-time PCR and ELISA results confirmed that iE-DAP reversed CSE-mediated effects on the mRNA levels and releases of IL-6, IL-8, TNF-α and IFN-γ by Leuk-1 cells. Taken together, our results indicated that NOD1 activation with iE-DAP could reverse CSE-mediated effects on NOD1 signaling in human oral mucosal epithelial cells.
Keywords: Cigarette smoke extract, NOD1 signal pathway, iE-DAP, cytokines, oral mucosal epithelial cells
Introduction
Cigarette smoking is well-known as a major risk factor in the development of various systemic diseases, including chronic obstructive pulmonary disease (COPD), cardiovascular diseases and cancer [1-4]. Cigarette smoking is also a recognized risk factor for oral disorders, such as oral cancer, oral leukoplakia and periodontal disease [5]. Cigarette smoke is a mixture of thousands of chemicals generated upon the burning or heating of tobacco leaves, many of which are cytotoxic, mutagenic, teratogenic or oncogenic [6].
In oral cavity, the epithelium is the first line of defense that encounters cigarette smoke and pathogens. These epithelial cells not only serve as a physical barrier, but also actively respond to microbes by producing cytokines, chemokines, inflammatory mediators, as well as antimicrobial peptides. Previous studies show oral epithelial cells can express pattern recognition receptors (PRR), such as Toll-like receptors (TLRs) and NOD-like receptors (NODs), which “sense” microorganisms and cellular damage by recognizing specific molecules through pathogen-associated pattern recognition (PAMP) and danger-associated pattern recognition (DAMP) [7-9].
As one of the best characterized members in NLR family, nucleotide-binding oligomerization domain 1 (NOD1) recognizes breakdown products of peptidoglycan (PGN). Once activated, NOD1 undergoes a conformational modification that allows the recruitment and activation of receptor-interacting protein 2 (RIP2), resulting in nuclear factor κB (NF-κB) activation and initiation of downstream gene transcription. A number of studies have demonstrated that NOD1 is a prominent molecule of innate immune and inflammatory response [7,8,10-13].
An early study has determined that NOD1 can be activated by γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP), a motif uniquely present in Gram-negative bacilli and particular Gram-positive bacteria [11]. Some previous reports also revealed that iE-DAP can induce innate and adaptive immune responses [14-16]. Our previous studies indicated that cigarette smoke extract (CSE) could suppress NOD1 expression and modulate NOD1 signal pathway in human oral mucosal epithelial cells [17,18]. Our results also confirmed that NOD1 agonist iE-DAP could reverse CSE-mediated effects on the levels of human β defensin (hBD)-1, -2, and -3, several key antimicrobial peptides [18]. Herein we hypothesize that iE-DAP could abrogate the inhibition of NOD1 expression by CSE exposure, thus to reverse the effects of CSE on some downstream cytokines of NOD1 signal pathway. In the present study, we investigated the regulatory role of iE-DAP in effects of CSE on NOD1 signal pathway in human oral mucosal epithelial cells.
Materials and methods
Reagents
Keratinocyte Serum-Free Medium (K-SFM) was purchased from GIBCO (Invitrogen, Carlsbad, CA, USA). Thiazolyl blue tetrazolium bromide (MTT) was purchased from Sigma (St. Louis, MO, USA). Phosphatase inhibitor cocktail was purchased from Roche (Mannheim, Germany), protease inhibitor cocktail was purchased from Fermentas UAB (Vilnius, Lithuania) and protein assay reagent and an enhanced chemiluminescent (ECL) kit were purchased from Pierce (Rockford, IL, USA). The following primary antibodies were used: rabbit anti-NOD1 antibody, mouse anti-RIP2 antibody and rabbit anti-P-NF-kB (p-p65) antibody were purchased from Abcam (Cambridge, UK) and rabbit anti-GAPDH antibody was purchased from Cell Signaling (Danvers, MA, USA). The peptidoglycan (PNG)-like molecules iE-DAP (γ-D-glutamyl-meso-diaminopimelic acid) and the negative control compounds iE-Lys (γ-D-glutamyl-Lysine) (all with endotoxin levels < 0.125 EU/ml) were purchased from InvivoGen (San Diego, California, USA). TRIzol reagent was purchased from Invitrogen (Carlsbad, CA, USA). RevertAid First Strand cDNA synthesis kit was purchased from Thermo (Waltham, MA, USA), and SYBR Green PCR Master Mix was purchased from Roche (Mannheim, Germany). Enzyme-linked immunosorbent assay (ELISA) kits were purchased from R&D Systems (Minneapolis, MN, USA).
Cell culture
Immortalized human oral mucosal epithelial (Leuk-1) cell line was a generous gift from Professor Li Mao at Department of Oncology and Diagnostic Sciences, University of Maryland Dental School, Baltimore, MD. The cells were expanded and passaged in K-SFM. This medium was supplemented with BPE (25 mg/ml), epidermal growth factor (0.2 ng/ml), CaCl2 (0.4 mM). Fresh media were replenished every 2-3 days. The passaged cells were cultured in 37°C humidified air incubators with 5% CO2. Cells were routinely cultured until 70% confluency, and trypsinized with 0.25% trypsin/0.02 EDTA solution.
Preparation of cigarette smoke extract (CSE)
Research-grade cigarettes (3R4F) were obtained from the Tobacco Research Institute, University of Kentucky (Lexington, KY, USA). CSE was prepared as previously described. Four cigarettes were bubbled through 40 ml of cell growth medium, and the stock solution (denoted 100% CSE) was diluted and used to stimulate cells at different final concentrations. CSE was freshly made, diluted and applied to cell cultures within 30 min of preparation.
Cell viability assay
Cell viability was determined by MTT assay as previously described [18]. Leuk-1 cells were cultured in 96-well plates at a density of 1×105 cells/ml and then were treated with various concentrations (0, 1%, 2%, 4%, 8%, 16% and 32%) of CSE or with different concentrations (0, 0.1, 1, 10, 50, 100 and 1000 μg/ml) of NOD1 agonist iE-DAP for 24 h at 37°C and 5% CO2 atmosphere. Ten microlitres of MTT solution (5 mg/ml in phosphate buffered-saline) was added into wells and incubated at 37°C in a 5% CO2 atmosphere for 4 h. Then the medium was aspirated, and 150 microlitres of DMSO was added and the plate was shaken to dissolve the formazan crystals. Absorbance was determined by using a multiplate reader (Bio-Rad 680, Hercules, CA, USA) at a wavelength of 570 nm.
NOD1 activation assay
NOD1 activation assay was performed as previously described [15]. Briefly, Leuk-1 cells were treated with various concentrations (0, 0.1, 1, 10, 50, and 100 μg/ml) of iE-DAP for 24 h. Then the total cellular protein was extracted and Western blotting was conducted. The relative level of NOD1 was determined by being normalized to that of GAPDH.
CSE and iE-DAP treatment
Leuk-1 cells were prepared as detailed above and the cell cultures were divided into four groups. And one half was treated with 4% CSE, while another half untreated. After 24 h, the medium was discarded and cells were then either treated with 50 μg/ml iE-DAP or 50 μg/ml iE-Lys (negative control) for another 24 h. Leuk-1 cells and the supernatant were subsequently collected for following experiments.
Western blotting
The cells were washed two times with ice-cold PBS and lysed in ice-cold lysis buffer containing 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, protease inhibitor cocktail, and phosphatase inhibitor cocktail. The lysates were incubated on ice for 30 min and centrifuged at 14,000 g for 10 min at 4°C to remove cell debris. The protein concentration was measured and the Western blotting was performed under the denaturing conditions. A sample of protein (20 mg) from the cell lysates was separated by SDS-PAGE in 10% polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) by wet electroblotting, which were further blocked in with 5% bovine serum albumin (BSA) in PBS-0.1% Tween 20 (PBST) for 1 h at room temperature (RT). Incubation with the primary antibodies (NOD1, RIP2, p-NF- kB diluted 1:1000 and GAPDH antibodies diluted 1:5000 with PBST containing 5% BSA) overnight at 4°C, and incubation with horseradish peroxidase-conjugated secondary antibodies (5,000-fold diluted with PBST containing 5% BSA) was conducted at RT for 1 h. Following washing with PBST, Immunostained protein bands were detected by using an enhanced chemiluminescence (ECL) assay kit and were visualized on FluorChem FC2 system (Cell Biosciences, Santa Clara, CA). Densitometric analyses of bands were performed using Image J software (http://rsb.info.nih.gov/ij/) and the data of target proteins were normalized to that of GAPDH.
Quantitative RT-PCR
Total RNA was extracted using TRIzol as described by the manufacturer according to the manufacturer’s instructions, and 2 μg of RNA was used to synthesize first-strand cDNA synthesis in 20 μl of reaction volume using the RevertAid First Strand cDNA Synthesis Kit according to the manufacturer’s protocol. The primers used for the PCR amplifications are listed as follows: IL-6: 5’-AAA TTC GGT ACA TCC TCG ACG G-3’, 5’-GGA AGG TTC AGG TTG TTT TCT GC-3’; IL-8: 5’-TCC TGA TTT CTG CAG CTC TG -3’, 5’-GTC CAC TCT CAA TCA CTC TCA G-3’; TNF-α: 5’-CTA TCT GGG AGG GGT CTT CC-3’, 5’-ATG TTC GTC CTC CTC ACA GG-3’; IFN-γ: 5’-ATC CCA TGG GTT GTG TGT TT-3’, 5’-CAA ACC GGC AGT AAC TGG AT-3’; GAPDH: 5’-GCA CCG TCA AGG CTG AGA AC-3’, 5’-TGG TGA AGA CGC CAG TGG A-3’. The cDNA was amplified using the SYBR Green PCR Master Mix and Real-time PCR analyses ware performed using an ABI Prism 7300 sequence detection system (Applied Biosystems). Amplification conditions were as follows: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, 58°C for 30 s, and 72°C for 30 s, followed by melting curve analysis, by which the specificity of primers was confirmed. The data were normalized to GAPDH and expressed as relative mRNA levels. Fold changes in gene expression were calculated by a comparative threshold cycle (Ct) method using the formula 2-(∆∆Ct).
Enzyme-linked immunosorbent assay (ELISA)
The supernatants of cell cultures were assessed for IL-6, IL-8, TNF-α and IFN-γ production by ELISA kits according to the manufacturer’s protocol.
Statistical analyses
Statistical analyses were performed using SPSS 15.0 (Chicago, IL). Data expressed as mean ± SE. Statistical significance (P<0.05) was determined using the unpaired t-test for differences between groups.
Results
Influences of CSE or iE-DAP on cell viability
The influences of various concentrations of CSE or iE-DAP on the viability of Leuk-1 cells were assessed by MTT assay. As Figure 1 shown, the treatment of cells with 1%~16% CSE for 24 h did not remarkably decrease the cell viability and the treatment of cells with 0.1 μg/ml~100 μg/ml iE-DAP for 24 h did not significantly alter the cell viability. However, the treatment with 32% CSE or 1000 μg/ml iE-DAP for 24 h prominently reduce the cell viability. Therefore, our results indicated that relatively low concentrations of CSE or iE-DAP had no significant influence on cell viability.
Figure 1.
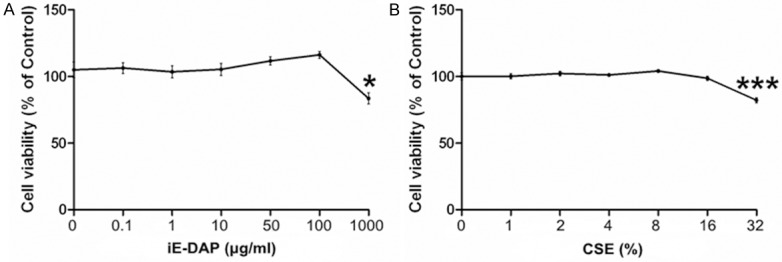
Influences of different concentrations of CSE or iE-DAP on the viability of Leuk-1 cells by MTT assay. A. Treatment of Leuk-1 cells with 50 μg/ml iE-DAP for 24 h did not significantly alter the cell viability. B. Treatment of Leuk-1 cells with 4% CSE for 24 h did not clearly reduce the cell viability. Relative cell viability was represented as means ± SE (n = 4). Statistical significance: *P < 0.05, ***P < 0.001, vs. control.
iE-DAP triggered NOD1 expression of leuk-1 cells in a dose-dependent manner
NOD1 expression in leuk-1 cells was gradually triggered by iE-DAP in a dose-dependent manner. Treatment of cells with 0.1, 1, 10, 50 and 100 μg/ml iE-DAP for 24 h clearly augmented NOD1 expression compared with the control. The level of NOD1 reached the peak at 24 h following the treatment with iE-DAP of 50 μg/ml concentration (Figure 2). Therefore 50 μg/ml was identified as the optimum concentration of iE-DAP which was used in the following experiments.
Figure 2.
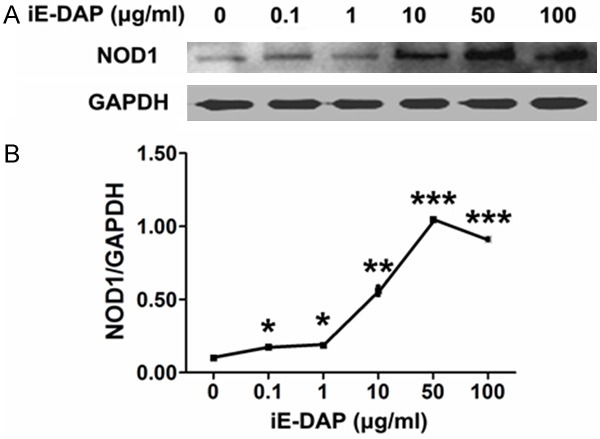
iE-DAP triggered NOD1 expression of leuk-1 cells in a dose-dependent manner. A. Representative immunoblot bands of NOD1. B. Density analyses of bands indicated that treatment of cells with 0.1, 1, 10, 50 and 100 μg/ml iE-DAP for 24 h clearly enhanced NOD1 expression. Following 50 μg/ml iE-DAP treatment for 24 h, NOD1 expression reached the peak level. Density data of bands were represented as means ± SE (n = 3). Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, vs. control.
iE-DAP reversed the suppressive effect of CSE on NOD1 expression and prevented the overactivation of RIP2 and P-NF-κB following CSE exposure
To determine whether NOD1 activation could modulate CSE-mediated expression changes of crucial molecules in NOD1 signal pathway, Leuk-1 cells were treated with 4% CSE for 24 h and 50 μg/ml iE-DAP for another 24 h. CSE treatment significantly inhibited NOD1 expression in Leuk-1 cells. Interestingly, the suppressive effect of CSE on NOD1 expression was prominently reversed following iE-DAP treatment (Figure 3A and 3B). We next examined whether NOD1 activation could regulate CSE-mediated expression changes of other crucial molecules in NOD1 signaling. CSE treatment remarkably increased RIP2 and P-NF-κB levels in Leuk-1 cells. Compared with the control, iE-DAP-treated cells also expressed significantly higher levels of RIP2 and P-NF-κB. However, the combination of CSE stimulation with iE-DAP treatment prevented the further enhancement of RIP2 and P-NF-κB levels (Figure 3A-D). Taken together, iE-DAP reversed the inhibitory effect of CSE on NOD1 expression and prevented the overactivation of RIP2 and P-NF-κB due to CSE exposure.
Figure 3.
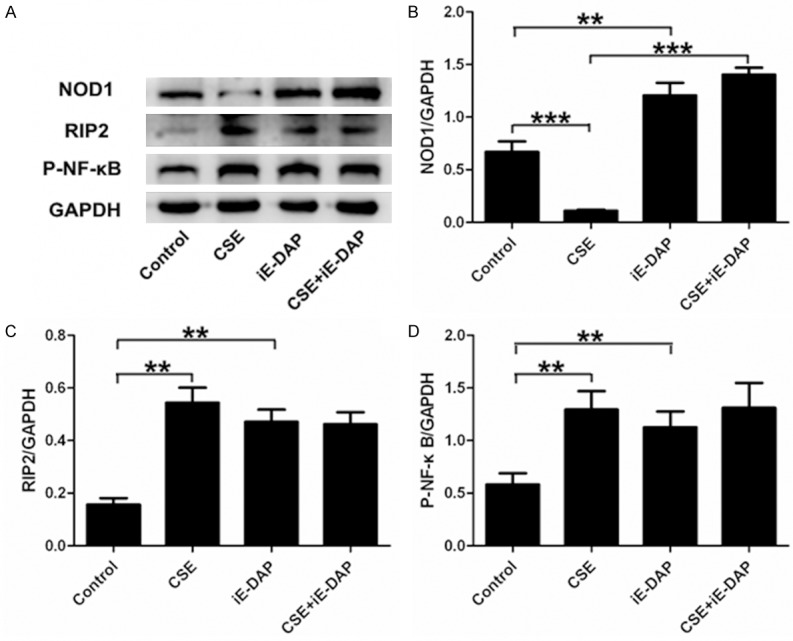
iE-DAP treatment reversed the inhibitory effect of CSE on NOD1 expression and prevented the overactivation of RIP2 and P-NF-κB following CSE exposure. A. Representative immunoblot bands of NOD1, RIP2 and P-NF-κB. B. Density analyses of bands indicated that CSE significantly inhibited NOD1 expression in Leuk-1 cells and iE-DAP treatment prominently reversed the suppressive effect of CSE on NOD1 expression. C, D. CSE treatment remarkably increased RIP2 and P-NF-κB levels in Leuk-1 cells. However, the combination of CSE exposure with iE-DAP treatment did not contribute to the further enhancement of RIP2 and P-NF-κB levels. Density data of bands were expressed as means ± SE (n = 3). Statistical significance: **P < 0.01, ***P < 0.001.
CSE upregulated levels of IL-6, IL-8 and TNF-α and downregulated IFN-γ level
In the present study, the qPCR data indicated that treatment with CSE for 24 h markedly upregulated mRNA levels of IL-6, IL-8 and TNF-α and downregulated IFN-γ mRNA level in Leuk-1 cells (Figure 4). The ELISA results also showed the same significant changes. The releases of IL-6, IL-8 and TNF-α clearly increased and IFN-γ release remarkably decreased in the supernatant of Leuk-1 cells following CSE treatment for 24 h (Figure 5).
Figure 4.
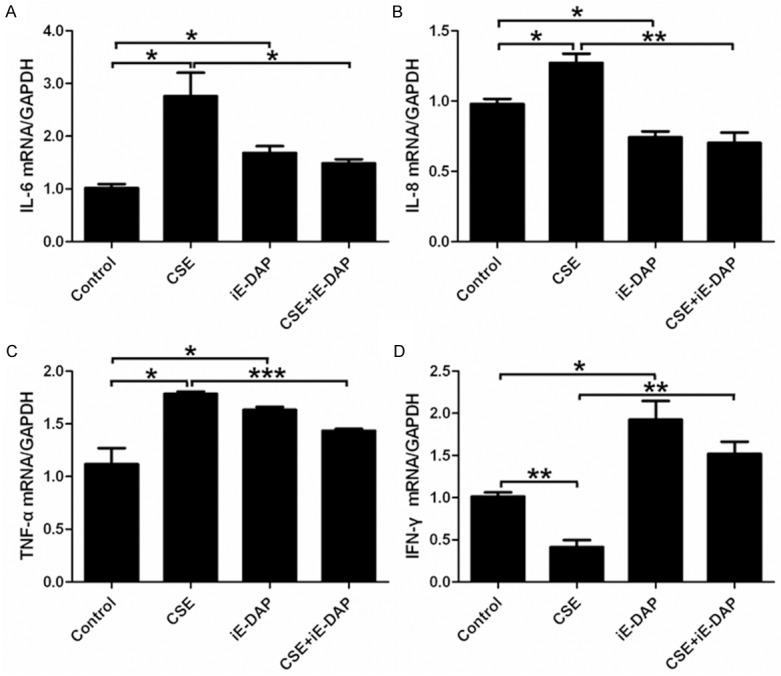
iE-DAP treatment reversed the effects of CSE on the mRNA levels of IL-6, IL-8, TNF-α and IFN-γ in Leuk-1 cells. A-C. Treatment of cells with iE-DAP significantly abrogated the induced effect of CSE on mRNA expression of IL-6, IL-8 and TNF-α. D. iE-DAP treatment remarkably reversed the inhibitory effect of CSE on IFN-γ mRNA level. The relative mRNA levels were expressed as means ± SE (n = 3). Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 5.
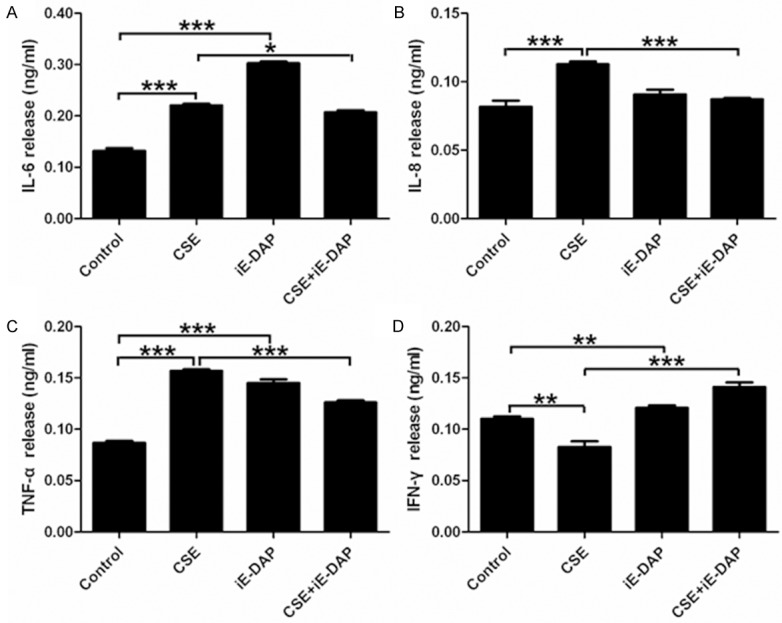
iE-DAP treatment reversed the impacts of CSE on the releases of IL-6, IL-8, TNF-α and IFN-γ from Leuk-1 cells. A-C. Treatment of Leuk-1 cells with iE-DAP conspicuously removed the activated effect of CSE on the releases of IL-6, IL-8 and TNF-α. D. Treatment of Leuk-1 cells with iE-DAP markedly abolished the suppressive effect of CSE on IFN-γ release. The ELISA data were expressed as means ± SE (n = 3). Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001.
iE-DAP enhanced the levels of IL-6, TNF-α and IFN-γ by Leuk-1 cells
Our results indicated that iE-DAP remarkably augmented the gene expression and release of IL-6, TNF-α and IFN-γ by Leuk-1 cells. However, iE-DAP markedly diminished mRNA level of IL-8 in Leuk-1 cells and did not conspicuously affect the production of IL-8 at protein level (Figures 4 and 5).
iE-DAP reversed CSE-mediated effects on the mRNA levels and releases of IL-6, IL-8, TNF-α and IFN-γ by Leuk-1 cells
To clarify effects of iE-DAP on CSE-mediated alteration of IL-6, IL-8, TNF-α and IFN-γ levels, Leuk-1 cells were treated with CSE and iE-DAP, either alone or in combination with each other. Our data indicated that treatment of Leuk-1 cells with iE-DAP significantly abrogated the induction of CSE on mRNA levels of IL-6, IL-8 and TNF-α (Figure 4A-C). Whereas iE-DAP treatment clearly eliminated the inhibition of CSE on IFN-γ mRNA level (Figure 4D). iE-DAP treatment markedly removed the activation of CSE on releases of IL-6, IL-8, TNF-α and abolished the suppression of CSE on IFN-γ release (Figure 5). In short, our current data showed that NOD1 agonist iE-DAP reserved CSE-mediated effects on IL-6, IL-8, TNF-α and IFN-γ levels.
Discussion
Many studies have demonstrated that various cell types including oral epithelial cells express NOD1, which plays an important role in innate immune and inflammatory response [7,8,19]. Some data in the literature confirmed that NOD1 agonist iE-DAP could augment NOD1 expression in a dose-dependent manner [15]. Our present results also supported the finding.
Our recent studies confirmed that CSE could inhibit NOD1 expression in oral mucosal epithelial cells [17,18]. Our data also suggested that NOD1 expression decreased in oral mucosa of smokers compared with that of nonsmokers [18]. In the present study, our results for the first time determined that iE-DAP could reverse the inhibitory effect of CSE on NOD1 expression in oral mucosal epithelial cells. We previously reported that the reduced NOD1 expression is significantly associated with oral squamous cell carcinoma (OSCC) progression [20]. All the evidences could link the suppressive effect of smoking on NOD1 expression to the occurrence and development of some oral diseases, while iE-DAP could potentially be used to treat and prevent these diseases in future.
Consistent to the results from previous studies, our present data showed that NOD1 stimulation with iE-DAP could increase RIP2 and P-NF-κB expression [13,15,21]. Previously, our results suggested that RIP2 and P-NF-κB expression increased in human oral mucosal epithelial cells following CSE exposure [17,18]. Our present results further indicated that NOD1 activation with iE-DAP prevented the overactivation of RIP2 and P-NF-κB due to CSE exposure. Moreover our current data showed that iE-DAP reserved effects of CSE on IL-6, IL-8, TNF-α and IFN-γ levels. Recently, we reported that the activation of NOD1 by iE-DAP could reversed the effects of CSE on hBD-1, -2, and -3 levels of oral mucosal epithelial cells [18]. NOD1 activation is subtly modulated by a negative autocrine feedback system, in which NOD1-regulated effectors concomitantly inhibit the downstream effects of NOD1 activation [22,23].
It has been confirmed that cigarette smoke or CSE exposure could increase the production of proinflammatory cytokines in aerodigestive cells [2,24]. Nevertheless smoking appears to inhibit innate immune and host defense against microbial infection while promoting or amplifying inflammatory reactions [24-27]. These effects of smoking could result in disruption of tissue homeostasis, duration of inflammatory response and diminished anti-microbial functions, which may partially explain the higher likelihood of smokers to develop colonization, invasion of pathogens, subsequent infection and chronic inflammation [28,29].
NOD1 activation could induce both innate immune and inflammatory response, characterized by the production of cytokines, chemokines and antimicrobial peptides. Among these downstream productions, some are proinflammatory, such as IL-6, IL-8, TNF-α and hBD-2, while others are not, such as IFN-γ and hBD-1. A short- or long-term exposure to cigarette smoke could result in the attenuation of innate immune and overactivation of inflammatory response. Taken these factors into account, it is especially crucial to keep the balance between innate immune and inflammatory response, which contributes to efficient clearance of pathogens, not to lead to excessive inflammatory damages.
Although effects of iE-DAP on cytokine production have been widely focused in the literature, study data are somewhat in conflict. It was reported that iE-DAP could activate various human epithelial cells to produce anti-microbial peptides, but not proinflammatory cytokines, such as IL-6 and IL-8 [7,8,19,30]. While it was also found that NOD1 activation induces the production of proinflammatory cytokines in human intestinal epithelial cells, dental pulp fibroblasts, adipocytes and trophoblast cells [8,11,15,31-33]. Our present study showed that iE-DAP could lead to the up-regulation of IL-6, TNF-α and IFN-γ in oral mucosal epithelial cells. However, iE-DAP downregulated mRNA level of IL-8 and did not increase the production of IL-8 in the present study. These differential results could be explained with different types of cell lines used in these studies, durations and concentrations of iE-DAP treatment.
It has been reported that iE-DAP has the potential as immunomodulators in particulate vaccine carriers [34]. A most recent study indicated that the preactivation of NOD1 with iE-DAP before bacterial challenge could suppress the inflammation and cancer lesion in the stomach of Mongolian gerbils in response to H. pylori [23]. The present results indicated that iE-DAP could antagonize CSE-mediated effects on NOD1 signaling and downstream effectors to a certain extent. These findings provide a new insight into regulatory effects of iE-DAP on smoking-induced dysfunction of innate immune and inflammatory response. Therefore, iE-DAP could be potentially used for the improvement of treatment and prevention of some smoking-related aerodigestive diseases in future.
Acknowledgements
This work was supported by the National Natural Scientific Foundation of China (No. 81070839, No. 81273121 and No. 81400521), Jiangsu Province’s Outstanding Medical Academic Leader program (No. LJ201110), the Key Project of Science and Technology Department of Jiangsu Province (No. BL2014018) and the Science and Technology Development Program of Nanjing (No. 201402033). We would like to thank Professor Li Mao (School of Dentistry, University of Maryland, USA) for his kind provision of oral mucosal epithelial (Leuk-1) cell line and Professor Wantao Chen (The Ninth People’s Hospital, School of Stomatology, Shanghai Jiao Tong University, China) for his kind help to our experiment.
Disclosure of conflict of Interest
None.
References
- 1.Zhang L, Ren JW, Wong CC, Wu WK, Ren SX, Shen J, Chan RL, Cho CH. Effects of cigarette smoke and its active components on ulcer formation and healing in the gastrointestinal mucosa. Curr Med Chem. 2012;19:63–69. doi: 10.2174/092986712803413926. [DOI] [PubMed] [Google Scholar]
- 2.Shen Y, Tian P, Li D, Wu Y, Wan C, Yang T, Chen L, Wang T, Wen F. Chrysin suppresses cigarette smoke-induced airway inflammation in mice. Int J Clin Exp Med. 2015;8:2001–2008. [PMC free article] [PubMed] [Google Scholar]
- 3.Hu JP, Zhao XP, Ma XZ, Wang Y, Zheng LJ. Effects of cigarette smoke on aerobic capacity and serum MDA content and SOD activity of animal. Int J Clin Exp Med. 2014;7:4461–4465. [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J, Juedes N, Narayan VM, Yue B, Rockwood AL, Palma NL, Patel JM. A cellular model to mimic exhaled cigarette smokeinduced lung microvascular endothelial cell injury and death. Int J Clin Exp Med. 2010;3:223–232. [PMC free article] [PubMed] [Google Scholar]
- 5.Wang WM, Ye P, Qian YJ, Gao YF, Li JJ, Sun FF, Zhang WY, Wang X. Effects of whole cigarette smoke on human beta defensins expression and secretion by oral mucosal epithelial cells. Tob Induc Dis. 2015;13:3. doi: 10.1186/s12971-015-0029-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hecht SS. Cigarette smoking: cancer risks, carcinogens, and mechanisms. Langenbecks Arch Surg. 2006;391:603–613. doi: 10.1007/s00423-006-0111-z. [DOI] [PubMed] [Google Scholar]
- 7.Sugawara Y, Uehara A, Fujimoto Y, Kusumoto S, Fukase K, Shibata K, Sugawara S, Sasano T, Takada H. Toll-like receptors, NOD1, and NOD2 in oral epithelial cells. J Dent Res. 2006;85:524–529. doi: 10.1177/154405910608500609. [DOI] [PubMed] [Google Scholar]
- 8.Uehara A, Fujimoto Y, Fukase K, Takada H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol. 2007;44:3100–3111. doi: 10.1016/j.molimm.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 9.Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, Devera ME, Liang X, Tor M, Billiar T. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 10.Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, Ogura Y, Nunez G. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–27831. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- 11.Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, Ogura Y, Kawasaki A, Fukase K, Kusumoto S, Valvano MA, Foster SJ, Mak TW, Nunez G, Inohara N. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4:702–707. doi: 10.1038/ni945. [DOI] [PubMed] [Google Scholar]
- 12.Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Nunez G, Inohara N. A critical role of RICK/RIP2 polyubiquitination in Nodinduced NF-kappaB activation. EMBO J. 2008;27:373–383. doi: 10.1038/sj.emboj.7601962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Wu J, Xin Z, Wu X. Aspergillus fumigatus triggers innate immune response via NOD1 signaling in human corneal epithelial cells. Exp Eye Res. 2014;127:170–178. doi: 10.1016/j.exer.2014.07.025. [DOI] [PubMed] [Google Scholar]
- 14.Cardenas I, Mulla MJ, Myrtolli K, Sfakianaki AK, Norwitz ER, Tadesse S, Guller S, Abrahams VM. Nod1 activation by bacterial iEDAP induces maternal-fetal inflammation and preterm labor. J Immunol. 2011;187:980–986. doi: 10.4049/jimmunol.1100578. [DOI] [PubMed] [Google Scholar]
- 15.Zhou S, Yu P, Guan L, Xing A, Liu S, Xiong Q, Peng B. NOD1 expression elicited by iE-DAP in first trimester human trophoblast cells and its potential role in infection-associated inflammation. Eur J Obstet Gynecol Reprod Biol. 2013;170:318–323. doi: 10.1016/j.ejogrb.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 16.Lappas M. NOD1 and NOD2 regulate proinflammatory and prolabor mediators in human fetal membranes and myometrium via nuclear factor-kappa B. Biol Reprod. 2013;89:14. doi: 10.1095/biolreprod.113.110056. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Qian YJ, Zhou Q, Ye P, Duan N, Huang XF, Zhu YN, Li JJ, Hu LP, Zhang WY, Han XD, Wang WM. Caspase-12 silencing attenuates inhibitory effects of cigarette smoke extract on NOD1 signaling and hBDs expression in human oral mucosal epithelial cells. PLoS One. 2014;9:e115053. doi: 10.1371/journal.pone.0115053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian YJ, Wang X, Gao YF, Duan N, Huang XF, Sun FF, Han XD, Wang WM. Cigarette Smoke Modulates NOD1 Signal Pathway and Human beta Defensins Expression in Human Oral Mucosa. Cell Physiol Biochem. 2015;36:457–473. doi: 10.1159/000430112. [DOI] [PubMed] [Google Scholar]
- 19.Uehara A, Takada H. Synergism between TLRs and NOD1/2 in oral epithelial cells. J Dent Res. 2008;87:682–686. doi: 10.1177/154405910808700709. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Jiang W, Duan N, Qian Y, Zhou Q, Ye P, Jiang H, Bai Y, Zhang W, Wang W. NOD1, RIP2 and Caspase12 are potentially novel biomarkers for oral squamous cell carcinoma development and progression. Int J Clin Exp Pathol. 2014;7:1677–1686. [PMC free article] [PubMed] [Google Scholar]
- 21.Swaan PW, Bensman T, Bahadduri PM, Hall MW, Sarkar A, Bao S, Khantwal CM, Ekins S, Knoell DL. Bacterial peptide recognition and immune activation facilitated by human peptide transporter PEPT2. Am J Respir Cell Mol Biol. 2008;39:536–542. doi: 10.1165/rcmb.2008-0059OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe T, Asano N, Fichtner-Feigl S, Gorelick PL, Tsuji Y, Matsumoto Y, Chiba T, Fuss IJ, Kitani A, Strober W. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest. 2010;120:1645–1662. doi: 10.1172/JCI39481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suarez G, Romero-Gallo J, Piazuelo MB, Wang G, Maier RJ, Forsberg LS, Azadi P, Gomez MA, Correa P, Peek RM Jr. Modification of Helicobacter pylori Peptidoglycan Enhances NOD1 Activation and Promotes Cancer of the Stomach. Cancer Res. 2015;75:1749–1759. doi: 10.1158/0008-5472.CAN-14-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Semlali A, Witoled C, Alanazi M, Rouabhia M. Whole cigarette smoke increased the expression of TLRs, HBDs, and proinflammory cytokines by human gingival epithelial cells through different signaling pathways. PLoS One. 2012;7:e52614. doi: 10.1371/journal.pone.0052614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eddleston J, Lee RU, Doerner AM, Herschbach J, Zuraw BL. Cigarette smoke decreases innate responses of epithelial cells to rhinovirus infection. Am J Respir Cell Mol Biol. 2011;44:118–126. doi: 10.1165/rcmb.2009-0266OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Modestou MA, Manzel LJ, El-Mahdy S, Look DC. Inhibition of IFN-gamma-dependent antiviral airway epithelial defense by cigarette smoke. Respir Res. 2010;11:64. doi: 10.1186/1465-9921-11-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thimmulappa RK, Gang X, Kim JH, Sussan TE, Witztum JL, Biswal S. Oxidized phospholipids impair pulmonary antibacterial defenses: evidence in mice exposed to cigarette smoke. Biochem Biophys Res Commun. 2012;426:253–259. doi: 10.1016/j.bbrc.2012.08.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feldman C, Anderson R. Cigarette smoking and mechanisms of susceptibility to infections of the respiratory tract and other organ systems. J Infect. 2013;67:169–184. doi: 10.1016/j.jinf.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Mahanonda R, Sa-Ard-Iam N, Eksomtramate M, Rerkyen P, Phairat B, Schaecher KE, Fukuda MM, Pichyangkul S. Cigarette smoke extract modulates human beta-defensin-2 and interleukin-8 expression in human gingival epithelial cells. J Periodontal Res. 2009;44:557–564. doi: 10.1111/j.1600-0765.2008.01153.x. [DOI] [PubMed] [Google Scholar]
- 30.Uehara A, Sugawara Y, Kurata S, Fujimoto Y, Fukase K, Kusumoto S, Satta Y, Sasano T, Sugawara S, Takada H. Chemically synthesized pathogen-associated molecular patterns increase the expression of peptidoglycan recognition proteins via toll-like receptors, NOD1 and NOD2 in human oral epithelial cells. Cell Microbiol. 2005;7:675–686. doi: 10.1111/j.1462-5822.2004.00500.x. [DOI] [PubMed] [Google Scholar]
- 31.Hirao K, Yumoto H, Takahashi K, Mukai K, Nakanishi T, Matsuo T. Roles of TLR2, TLR4, NOD2, and NOD1 in pulp fibroblasts. J Dent Res. 2009;88:762–767. doi: 10.1177/0022034509341779. [DOI] [PubMed] [Google Scholar]
- 32.Lee YY, Chan CH, Hung SL, Chen YC, Lee YH, Yang SF. Up-regulation of nucleotide-binding oligomerization domain 1 in inflamed human dental pulp. J Endod. 2011;37:1370–1375. doi: 10.1016/j.joen.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 33.Zhou YJ, Zhou H, Li Y, Song YL. NOD1 activation induces innate immune responses and insulin resistance in human adipocytes. Diabetes Metab. 2012;38:538–543. doi: 10.1016/j.diabet.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Wischke C, Mathew S, Roch T, Frentsch M, Lendlein A. Potential of NOD receptor ligands as immunomodulators in particulate vaccine carriers. J Control Release. 2012;164:299–306. doi: 10.1016/j.jconrel.2012.06.034. [DOI] [PubMed] [Google Scholar]