

Abstract
Notch3 plays an important role in differentiation, migration and signal transduction of vascular smooth muscle cells (VSMCs). In this study, we used RNA interference (RNAi) technique to investigate the effect of knocking down the expression of the NOTCH3 gene in VSMCs on the phenotype determination under pathologic status. Real-time PCR and Western Blot experiments verified the expression levels of Notch3 mRNA and protein were reduced more than 40% and 50% in the NOTCH3 siRNA group. When the expression of Notch3 was decreased, the proliferation, apoptosis and immigration of VSMCs were enhanced compared to control groups (P < 0.01). NOTCH3 siRNA VSMCs observed using confocal microscopy showed abnormal nuclear configuration, a disorganized actin filament system, polygonal cell shapes, and decreasing cell sizes. Additionally, knocking down the expression of NOTCH3 may evoke the CASR and FAK expression. In Conclusion, interfering with the expression of NOTCH3 causes VSMCs to exhibit an intermediate phenotype. CaSR and FAK may be involved in the Notch3 signaling pathway.
Keywords: VSMCs, Notch3, RNAi, phenotype switch
Introduction
The Notch signaling pathway is an evolutionarily conserved intercellular signaling system that plays a central role during vascular development and in the physiology of vertebrates [1]. The Notch family receptors of humans and rodents contain 4 highly conserved members (Notch1-Notch4), which are single-pass transmembrane proteins consisting of both an extracellular domain (ECD) and an intracellular domain (ICD) [2,3]. Among these, Notch3 is expressed primarily in vascular smooth muscle cells [4]. The Notch3 gene (NOTCH3) encodes a single-pass transmembrane receptor of 2321 amino acids with an extracellular domain containing 34 epidermal growth factor repeats (EGFR), each including six cysteine residues [5]. Mutations in NOTCH3 can cause cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), which is the most common hereditary vascular dementia [6].
Notch3-null mice are viable and fertile, indicating that Notch3 is not required in embryonic development. However, Notch3 is necessary for differentiation of VSMCs and for acquisition of arterial identity [7,8]. In addition, it has been reported that genetically engineered mice without Notch3 have prominent small artery structural defects, as a result of impaired differentiation and maturation of VSMCs [8].
It is well known that there are two phenotypes of VSMCs: synthetic (dedifferentiated) and contractile (differentiated) [9]. Synthetic type switching of VSMCs is characterized by the loss of VSMC contractile genes and this step is believed to be critical for facilitating abnormal VSMC proliferation, migration and extracellular matrix synthesis [10,11]. Unlike skeletal and cardiac muscle cell lineages, VSMCs retain their capacity to proliferate and modulate their phenotype during postnatal development. The Notch pathway may also be involved in these steps. Several studies have characterized that the expression of several Notch pathway components, Notch3 included, plays a role in vascular injury [12]. Morrow et al. demonstrated that cyclic mechanical strain could increase the expression of NOTCH1 and NOTCH3 in VSMCs, and as a result, inhibit the proliferation of VSMCs while increasing apoptosis [13,14].
Several signaling pathways and transcription factors are important in dictating the phenotypic state (i.e., proliferative versus contractile) of VSMCs. Notch receptors were shown to inhibit VSMC differentiation in vitro through CBF-1/RBP-Jk-dependent mechanisms by either positively or negatively regulating the expression of VSMC-restrictive genes, such as smooth muscle-actin [SMA], calponin, smooth muscle myosin heavy chain [SM-MHC], and smoothelin [14,15]. Hey2 repressed multiple transcriptional regulatory elements controlling the expression of VSMC contractile genes in VSMC [16]. Sweeney et al. [17] found that constitutive activation of NOTCH3 inhibited the phenotypic switch from a “contractile” to a “synthetic” phenotype, and this effect was reversed by inhibiting the transcriptional activity of CBF1/RBP-Jk. Morrow et al. [13] found that over-expression of NOTCH3 can promote VSMC proliferation and induce apoptosis. However, the extent to which Notch3-mediated pathways coordinate in the regulation of VSMC phenotypes is largely unknown.
In our study, we investigated the morphological and functional changes of VSMC upon knockdown of NOTCH3, and we assessed the relationship between phenotype switching of VSMCs and Notch3.
Materials and methods
Cells and cell culture
Primary human VSMCs, bought from Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, were harvested from human aortic arteries of organ donors who died in traffic accidents. All post mortem samples were collected within 24 h of death and all cell cultures were established immediately after obtaining the samples. Written informed consents were obtained from the donor (or the next of kin) for the use of this sample in research before the acquisition of the vessels. The following study was approved by the Ethical committee of the Military General Hospital of Beijing PLA, Beijing, China. VSMCs were cultured in DMEM (Invitrogen, Grand Island, NY, USA) containing FBS (Invitrogen) in 5% CO2 at 37°C. All experiments were performed on cells from passage 4 to 10.
Reagents
The reagents are listed in Table 1.
Table 1.
List of reagents
| Reagent | Vendor | Catalog No. |
|---|---|---|
| pAD/CMV/V5-DEST Vectors | Invitrogen | V493-20 |
| DMEM | Invitrogen | 10567-014 |
| FBS | Invitrogen | 16000-044 |
| Opti-Mem | Invitrogen | 11058-021 |
| PBS | Sigma | P3326-10UG |
| TRIzol | Invitrogen | 15596-026 |
| PureLink DNAse set | Invitrogen | 12185010 |
| PureLink RNA mini kit | Invitrogen | 12183018A |
| High-Capacity cDNA Reverse Transcription kit | Applied Biosystems | 4368814 |
| Power SYBR green Mastermix | Applied Biosystems | 4368702 |
| BCA protein assay | Pierce | 23227 |
| Transwell | Costar, Corning | 3422 |
| Amersham ECL Plus Western Blotting Detection reagents | GE Healthcare Santa Cruz Biotech | RPN2132 |
| Polyvinylidene difluoride membranes | Bio-Rad | 162-0174 |
| NuPAGE Novex 4-12% bis-Tris gels | Invitrogen | NP0322BOX |
| Cell Counting Kit-8 | Beyotime Inst Biotech | C0038 |
| Annexin V-PE Apoptosis kit | BD Biosciences | 559763 |
| IGF-1 | Sigma | I1271 |
RNAi-based NOTCH3 knockdown
To generate adenovirus transduction particles, a set of 4pAd/CMV/V5-DEST vectors encoding siRNA targeting the NOTCH3 gene (GenBank accession number NM_000435.2, mRNA, 8089 bp) and a non-target siRNA control vector (TRC1 library, Sigma-Aldrich, St. Louis, MO, USA) were respectively co-transfected into HEK293A cells (Invitrogen) along with packaging plasmid pDONR221 (Invitrogen). VSMCs were seeded onto 6 cm plates (6 × 105 cells per plate) 24 h before transduction. Viral transduction was carried out using medium containing adenoviruses particles. The cells were fed with fresh complete medium 24 h later. The transducted VSMCs were selected 48 h after transduction until the end of experimentation. The effect of NOTCH3 silencing was assessed by western blot and quantitative PCR (qPCR). Stable cell lines created with two vectors named pAD-EGFP-Notch3-1 and pAD-EGFP-Notch3-3 showed significant reduction of Notch3 protein expression. Final experiments were performed using stable cell lines generated with a pAD-EGFP-Notch3-1 construct. The details were described in an earlier study [18].
Cell grouping
VSMCs were divided into 5 treatment groups. The first one was Non-Transfected group, which was as normal control. The second one was the Control siRNA group, which was transfected with a non-target control siRNA vector as negative control. The third was the NOTCH3 siRNA group, which was transfected with a pAD-EGFP-Notch3-1 construct and had decreased NOTCH3 expression level. The fourth group was normal VSMCs cultured with Insulin-like growth factor-1 (IGF-1, 100 μg/L), which was named IGF-1 group. The last one was NOTCH3 siRNA VSMCs cultured with IGF-1, named as NOTCH3 siRNA/IGF-1 group.
Analysis of gene expression
VSMCs were lysed with TRIzol (Invitrogen) and total RNA was extracted using the PureLink® RNA Mini Kit including DNAse treatment (Invitrogen). cDNA was synthesized with the cDNA Synthesis Kit (TaKaRa, Dalian, China). SYBR green qPCR Mastermix (Dongsheng Biotech, Guangzhou, China) with a Light Cycler Nano Real-Time-PCR system (Roche, Basel, Switzerland) was used for amplification. The housekeeping gene hACTB served as an internal control and the ΔΔCt method was used to calculate relative mRNA expression levels [18].
The respective primer sequences are shown in the Supplemental Material (Table S1).
Cell proliferation assay
Cell proliferation ratio was determined using WST-8 dye and the Cell Counting Kit-8 (Beyotime, Nantong, China). Briefly, 2 × 105 cells/well were seeded in a 6-well flat-bottomed plate, grown at 37°C for 24 h, and then placed in serum-starved conditions for 24 h. From 24 h to 6 d, 10 μL WST-8dye was added to each well of different groups every 24 h. Cells were incubated at 37°C for 3 h and the absorbance was determined at 450 nm using a micro-plate reader.
Western blot analysis
After total cellular Protein was isolated, protein concentrations were determined using the standard BCA protein assay (Pierce, Rockford, IL, USA). Equal amounts of protein were loaded in each lane. Proteins were separated by gel electrophoresis and then transferred onto polyvinylidene difluoride (PVDF) membranes. PVDF membranes were incubated in the primary and secondary antibody solution sequentially. Amersham ECL Plus Western Blotting Detection reagents (GE Healthcare, Rockford, IL, USA) were used for chemiluminescence. ImageJ analysis software (NIMH, Bethesda, MD, USA) was used to calculate the relative density of each band normalized to the internal control β-actin. Expression of those proteins that might be related to the phenotype of VSMCs or Notch3, such as α-Smooth Muscle Actin (α-SM-Actin), Smooth Muscle 22α (SM22α), osteopontin (OPN), and so on, were tested. The primary antibodies used are listed in Table 2.
Table 2.
List of Antibodies
| Reagent | Vendor | Catalog No. |
|---|---|---|
| Rabbit anti-Notch3 | Abcam | Ab60087 |
| TRITR-Phalloidine | Sigma-Aldrich | P1951 |
| FITC-Phalloidine | Sigma-Aldrich | P5282 |
| Mouse anti-β-Actin | Sigma | sc-47778 |
| Mouse anti-α-SM-Actin | Sigma-Aldrich | A2228 |
| Mouse anti-SM22α | Abcam | Ab14106 |
| Rabbit anti-OPN | Abcam | Ab8448 |
| Mouse anti-CaSR | Sigma | SAB1405561 |
| Rabbit anti-FAK | Sigma | SAB4300665 |
Apoptosis assay
The detection of apoptosis was performed using the Annexin V-PE Apoptosis kit (BD Biosciences, Franklin Lake, NY, USA). VSMCs were washed, trypsinized, and pelleted. The supernatant was removed, and cells were resuspended in the provided staining buffer containing the Annexin V-PE antibody. Analysis was carried out using flow cytometry (BD FACSCalibur, Franklin Lake, NY, USA). Data analysis was performed with Cell Quest software.
Cell morphology and immunocytochemistry
VSMCs were fixed with paraformaldehyde for analyzing the expression of cell surface Notch3 and cell morphology. Fixed cells were blocked with 3% bovine serum albumin in PBS for 30 min and incubated with Notch3 primary antibodies (Abcam, Cambridge, UK), followed by Cy3-labeled secondary antibody incubation. After being washed five times in PBS, VSMCs were stained by FITC or TRITC labeled phalloidin to show the expression of Actin, followed by DAPI staining to show cell nuclei. Cell images were analyzed and captured with a FluoView 1000 laser scanning confocal microscope (Olympus, Tokyo, Japan).
Transwell migration assay
One day before the migration assay, VSMCs were cultured in DMEM containing 0.1% serum. On the day of the experiment, cells were trypsinized and seeded onto the Transwell inserts (30,000 cells/insert, 6.5-mm diameter) (Transwell, Costar, Corning, NY, USA). Assays were stopped 48 h later and inserts were washed three times in PBS. Migrated cells were stained by Regent B of crystal violet stain kit (GenMed, Minneapolis, MN, USA) for 20 min. We used a light microscope to enumerate the number of stained cells in random fields within each Transwell Insert. Then the stained cells were lysed in 1.5 mL Regent C and 100 μL was transferred to a 96-well plate for absorbance measurement at 570 nm by a micro-plate reader.
Statistics analysis
Each experiment was repeated at least three times. Quantitative PCR data, cell proliferation and migration assays, and Western Blot data were compared using one-way ANOVA with LSD post hoc correction as appropriate. P-values < 0.05 were considered statistically significant.
Results
NOTCH3 gene and protein expression were reduced by RNAi
With the expression of the NOTCH3 in the control siRNA group defined as reference value, the relative NOTCH3 expression of the non-transfected group and the NOTCH3 siRNA group were 1.003 ± 0.082 and 0.571 ± 0.014, respectively. Thus, NOTCH3 expression was reduced more than 40% in the NOTCH3 siRNA group (P < 0.01), as detected by quantitative PCR. The details were described in an earlier study [18].
The mRNA data were corroborated by evaluation of Notch3 protein levels. Expression of β-actin was used as internal control. The relative expression level of Notch3 protein in the non-transfected group was 0.235 ± 0.013; in the control siRNA group it was 0.218 ± 0.001, in the NOTCH3 siRNA group it was 0.124 ± 0.004, and in the IGF-1 group it was 0.214 ± 0.010, and in the NOTCH3 siRNA/IGF-1 group it was 0.122 ± 0.010. NOTCH3 expression decreased by more than 50% in cells transfected with NOTCH3 siRNA sequences, compared to controls (P < 0.01). On the other hand, culture with IGF-1 did not alter Notch3 protein levels of normal cells nor NOTCH3 siRNA cells (P > 0.05) (Figure 1A).
Figure 1.
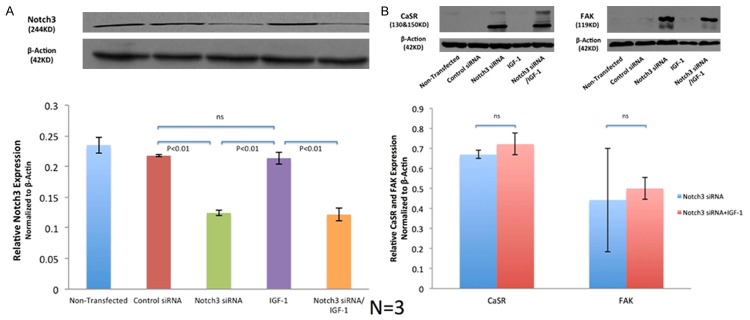
Notch3 and other proteins expression in different VSMCs Groups. A. Protein levels of Notch3 were compared among 5 groups, with β-actin as an internal control. The expression of Notch3 was lowest in NOCTH3 siRNA and NOTCH3 siRNS/IGF-1 groups. IGF-1 group did not show significant different Notch3 expression level, when comparing with the control. B. Significantly higher expression of CaSR and FAK was detected in in NOTCH3 siRNA with or without IGF-1. Relative expression levels between these two groups were not significant. This expression was not observed in the other three groups.
The expression of other relevant proteins varies in different groups
The two control groups had the highest expression of α-SM-Actin and SM22α, without any OPN expression detected. The relative expression of α-SM-Actin in IGF-1 group was the lowest (0.118 ± 0.054), and its expression of OPN was the highest (0.258 ± 0.041), if compared with other groups (P < 0.05). We didn’t detect the expression of SM22α in this group. About the NOTCH3 siRNA and NOTCH3 siRNA/IGF-1 VSMCs, the expression of α-SM-Actin and SM22α were moderate and also showed significant different if compared to controls and IGF-1 groups (P < 0.05). And OPN was also detected in the two groups, though the relative contents were much lower than IGF-1 group (P < 0.05) (Table 3).
Table 3.
The expressions of VSMCs phenotype related proteins
| α-SM-Actin | SM22α | OPN | |
|---|---|---|---|
| Non-Transfected | 0.479 ± 0.043 | 0.212 ± 0.039 | - |
| Control siRNA | 0.542 ± 0.078 | 0.262 ± 0.071 | - |
| NOTCH3 siRNA | 0.337 ± 0.066Δ,* | 0.116 ± 0.024Δ | 0.125 ± 0.017* |
| IGF-1 | 0.118 ± 0.054Δ | - | 0.258 ± 0.041 |
| NOTCH3 siRNA/IGF-1 | 0.289 ± 0.074Δ,* | 0.107 ± 0.030Δ | 0.156 ± 0.036* |
P < 0.05, compared with control groups.
P < 0.05, compared with IGF-1 group.
Surprisingly, in NOTCH3 siRNA VSMCs, significantly higher expression of CaSR and FAK was detected. This was also observed in NOTCH3 siRNA/IGF-1 VSMCs. Relative expression levels between two groups were not significant (P > 0.05), while the expression of CaSR and FAK was not detected in other three groups (Figure 1B).
NOTCH3 knockdown resulted in higher proliferation and apoptosis
At 144 h, the proliferation kinetics of NOTCH3 siRNA VSMCs was faster than in non-transfected and control siRNA cells (P < 0.01). Non-transfected VSMCs and control siRNA VSMCs had similar growth curves. VSMCs with NOTCH3 siRNA/IGF-1 showed the fastest proliferation kinetics, but was not significant different when compared to NOTCH3 siRNA VSMCs or IGF-1 VSMCs (Figure 2A).
Figure 2.
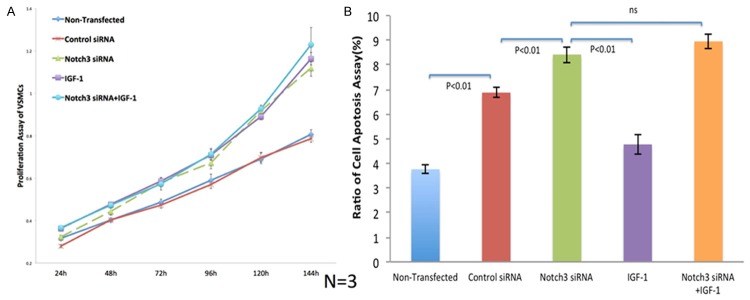
Proliferation and apoptosis of VSMCs in different groups. A. NOTCH3 siRNA/IGF-1 groups showed the fastest proliferation kinetics, but not significant different when compared to NOTCH3 siRNA or IGF-1 groups. Two control groups showed similar growth curves. B. The ratios of apoptosis and necrosis detected in NOTCH3 siRNA and NOTCH3 siRNA/IGF-1 groups were much higher than those in the other three groups.
At 72 h, the highest percentages of apoptosis and necrosis were in the NOTCH3 siRNA group with or without IGF-1 interfering, which was significantly different from other groups (P < 0.01) (Figure 2B).
NOTCH3 knockdown changed the migrating of VSMCs
There was a significant difference in migratory response between the non-transfected or control siRNA groups and the NOTCH3 siRNA, IGF-1 groups, or NOTCH3 siRNA and IGF-1 groups combined (P < 0.01). Although VSMCs cultured with IGF-1 showed slightly higher migration than NOTCH3 siRNA VSMCs, there was no significant difference among these three groups (Figure 3).
Figure 3.
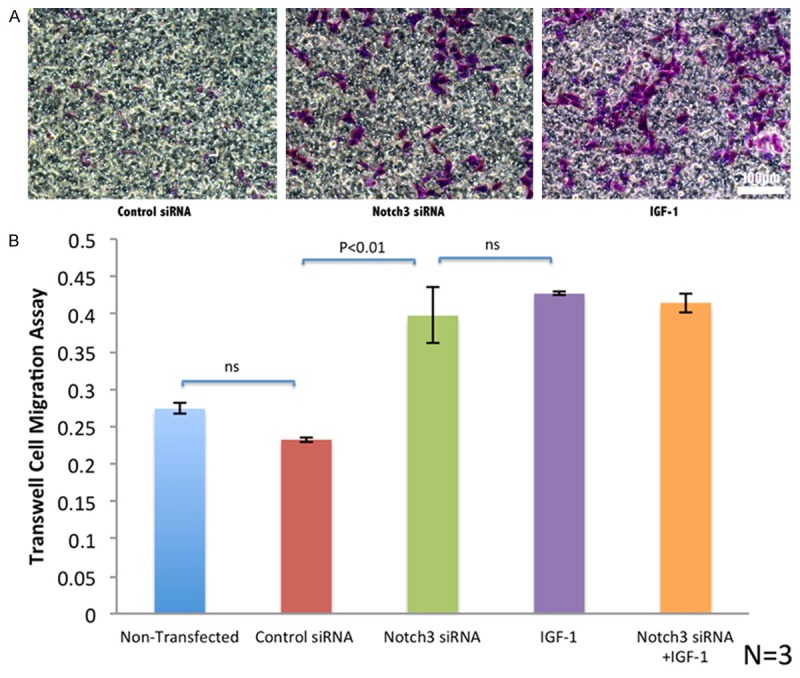
Stained VSMCs within each transwell insert under a light microscope. A. VSMCs (stained in purple) migrated into the transwell were detected under the microscope. While some migration was detected in the control, more migration was detected in NOTCH3 siRNA transwell and most in IGF-1 VSMCs. B. In vitro transwell assays indicated that the VSMC migration was higher in NOTCH3 siRNA and/or IGF-1 interfering than that in the control groups. The migration was very similar in all other three groups. The scale bar equals 100 µm.
Morphologic change after NOTCH3 knockdown
There was no difference in the morphology or cytoskeleton pattern between non-transfected and control siRNA VSMCs. Actin-phalloidin staining revealed normal actin filament patterning and cell shapes. Notch3-Cy3 staining suggested that Notch3 was expressed predominately on the cell membrane, as expected (Figure 4).
Figure 4.
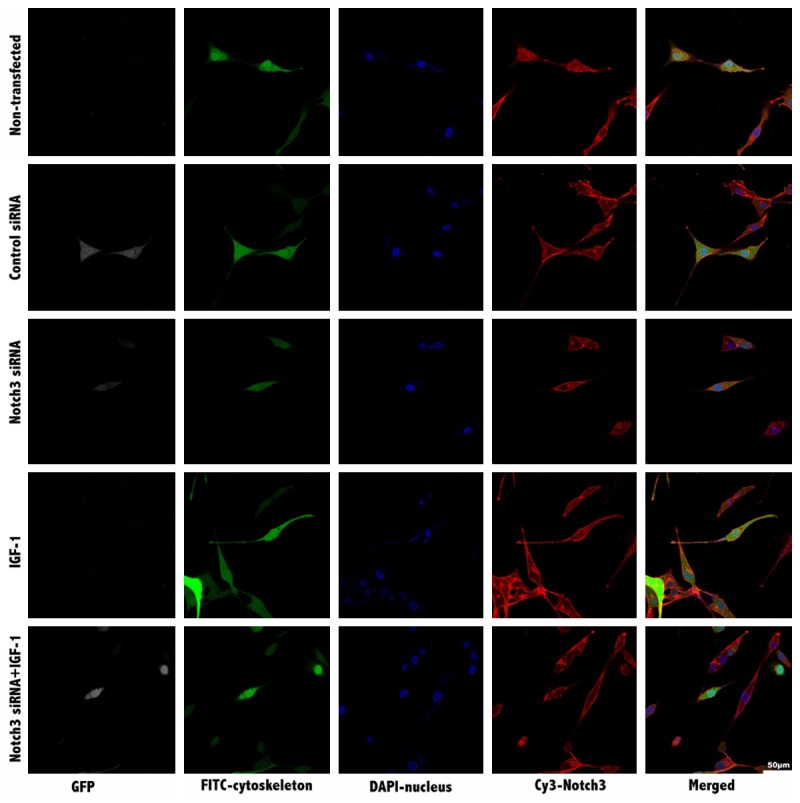
Morphology and structure analysis of VSMCs using confocal microscopy. GFP was shown in grey, FITC-labeled cytoskeleton was shown in green, Notch3 expression was staining in red. Nuclei were stained with DAPI in blue. Merged images at the same site are shown at the very right column. The scale bar equals 50 µm.
Actin-phalloidin staining in NOTCH3 siRNA VSMCs revealed abnormal nuclear configurations, disorganized actin filament distribution and polygonal cell shape. In addition to the decreased cell sizes and reduced expression of NOTCH3, some filaments of F-actin shrunk and even lost (Figure 4).
After cultured with IGF-1, the number of polymorphous cells significantly increased. This was accompanied by decreased intercellular gaps and increased cell sizes. Some cells became slender with extremely long tentacles. When observed under high magnification, more pseudopods with F-actin were observed and some filaments of F-actin became warped (Figure 4). These findings suggested that the increased migration of IGF-1 VSMCs may be at least partly related to the rearrangement of actin filaments. Notch3-Cy3 staining revealed similar expression levels of NOTCH3 with controls (Figure 4), which was consistent with the results of the western blot assay.
Discussion
Notch3 is expressed primarily in VSMCs and is required for arterial specification of VSMCs but not of endothelial cells. It is the first cell-autonomous regulator of arterial differentiation and maturation of VSMCs. In vivo studies have shown that Notch3−/− vessels lose their arterial phenotype, indicating that Notch3 is crucial for the phenotypic integrity of VSMCs [19]. The central component of the contractile apparatus in VSMCs is comprised of SM22α and α-SM-Actin. Some studies demonstrate that suppression of endogenous Notch3 receptors using siRNA significantly reduces endogenous levels of α-SM-actin and SM22α, indicating that basal Notch signaling promotes the contractile phenotype, but other studies have yielded conflicting results [20].
Mutations in NOTCH3 cause cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), including migraine with aura, ischemic attacks, cognitive impairment, mood disturbances and apathy [21]. CADASIL-associated mutations in NOTCH3 are located in the EGF-like repeats. Investigation of CADASIL mutant NOTCH3 shows that the majority of mutations do not change CBF1/JBP-Jk mediated classic Notch activation, so the pathological consequences of NOTCH3 mutations in CADASIL patients cannot be simply explained by loss-or gain-of-function in the classic Notch signaling pathway. This suggests that a novel Notch3-mediated signaling pathway may be present in VSMCs, or that cross-regulation of Notch3 to other signaling pathway(s) may play a critical role in VSMCs survival.
Therefore, it is of little surprise that the regulatory mechanisms overseeing Notch signaling are complex, interrelated, and apparently redundant. Additional layers of Notch regulation have recently been described in reviews, including soluble DSL ligand expression, cis-ligand expression, protein glycosylation and ubiquitination, regulation of ligand and Notch expression patterns [22-24].
In our study, we successfully knocked down the expression of NOTCH3 in VSMCs by RNA interference (RNAi). VSMC migration plays a critical role in the initiation and progression of intimal thickening in atherosclerotic lesions [25]. VSMCs acquire a synthetic phenotype leading to extensive migration, proliferation, and matrix synthesis that contributes to restenosis. Thus, prevention of pathological VSMC proliferation still remains a major clinical challenge, which underlines the need for new therapeutic strategies.
We found that reduced NOTCH3 expression promotes VSMC migration. VSMC migration promotes artery restenosis and vein graft failure. Actin-phalloidin staining in experimental VSMCs group showed abnormal nuclear configuration, a disorganized actin filament patterning and polygonal cell shapes. In our study, VSMCs with decreased NOTCH3 expression level with or without IGF-1 proliferated quicker and exhibited more pseudopods with F-actin, and some filaments of F-actin became warped, exhibiting different morphology from control VSMCs. Confocal microscopy revealed many heterocysts and polymorphous cells in the siRNA VSMC group.
Overall, the results described above indicated that knocking down the expression of NOTCH3 in VSMCs did not show typical contractile or synthetic phenotypes. Instead, they showed a more intermediate phenotype, suggesting Notch3 signaling is only one of the multiple pathways in the phenotype switching of VSMCs, or that NOTCH3 in our study was knocked down only by about 30-40%.
Unexpectedly, high expression of CaSR and FAK was detected in NOTCH3 siRNA VSMCs. In fact, we also detected proteins such as PCNA, c-Myc, c-Fos, PHLPP, and FilaminA, but their expression was not significantly different among groups (data not shown). Similar results were also observed in NOTCH3 siRNA/IGF-1 VSMCs. CaSR, an ion-sensing G protein-coupled receptor that is functionally expressed in the vasculature, couples to integrins, and in conjunction with intracellular calcium release, promotes cellular adhesion and migration in tumor cells [26]. Focal adhesions (FAs) are the cellular micro domains that mediate cell migration [27]. FAK plays a central role in regulating FA dynamics and cell motility [28,29]. The actions of FAK activation and FA dynamics have important consequences for VSMC motility, such as promoting migration of VSMCs [30]. NOTCH3 knockdown causes VSMCs to switch from contractile to synthetic, and promotes migration. Thus, FAK and CaSR may be potential therapeutic targets for atherosclerosis therapy and vein grafts.
A few limitations of this study need to be addressed. First, we investigated limited protein expression, morphology and filament distribution of VSMCs only in the groups mentioned in this study. Second, this study focused on phenotypic switching and migration of VSMCs, but other pathologic conditions (e.g., ischemia, hypoxia) were not examined. Future studies are needed to explore these topics. Finally, interactions between Notch and the microenvironment need to be taken into account, such as interactions with cytokines, ECM and other VSMC transcriptional regulators.
In summary, using RNA interference, we studied the outcomes of NOTCH3 knockdown in VSMCs. After interfering with the expression of NOTCH3, VSMCs exhibited an intermediate phenotype. NOTCH3 knockdown evoked the expression of CaSR and FAK, which may be useful for studying the Notch3 signaling pathway in detail.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Hofmann JJ, Iruela-Arispe ML. Notch signaling in blood vessels: who is talking to whom about what? Circ Res. 2007;100:1556–68. doi: 10.1161/01.RES.0000266408.42939.e4. [DOI] [PubMed] [Google Scholar]
- 2.Gridley T. Notch signaling in vascular development and physiology. Development. 2007;134:2709–2718. doi: 10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- 3.Iso T, Hamamori Y, Kedes L. Notch signaling in vascular development. Arterioscler Thromb Vasc Biol. 2003;23:543–553. doi: 10.1161/01.ATV.0000060892.81529.8F. [DOI] [PubMed] [Google Scholar]
- 4.Joutel A, Andreux F, Gaulis S, Domenga V, Cecillon M, Battail N, Piga N, Chapon F, Godfrain C, Tournier-Lasserve E. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest. 2000;105:597–605. doi: 10.1172/JCI8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cécillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 6.Joutel A, Vahedi K, Corpechot C, Troesch A, Chabriat H, Vayssière C, Cruaud C, Maciazek J, Weissenbach J, Bousser MG, Bach JF, Tournier-Lasserve E. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet. 1997;350:1511–5. doi: 10.1016/S0140-6736(97)08083-5. [DOI] [PubMed] [Google Scholar]
- 7.Krebs LT, Xue Y, Norton CR, Sundberg JP, Beatus P, Lendahl U, Joutel A, Gridley T. Characterization of Notch3-deficient mice: normal embryonic development and absence of genetic interactions with a Notch1 mutation. Genesis. 2003;37:139–143. doi: 10.1002/gene.10241. [DOI] [PubMed] [Google Scholar]
- 8.Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, Tournier-Lasserve E, Gridley T, Joutel A. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004;18:2730–5. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshinaga M, Sunagawa M, Shimada S, Nakamura M, Murayama S, Kosugi T. Argatroban, specific thrombin inhibitor, induced phenotype change of cultured rabbit vascular smooth muscle cells. Eur J Pharmacol. 2003;461:9–17. doi: 10.1016/s0014-2999(03)01293-7. [DOI] [PubMed] [Google Scholar]
- 10.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–9. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, Campos AH, Prince CZ, Mou Y, Pollman MJ. Coordinate Notch3-hairy-related transcription factor pathway regulation in response to arterial injury. Mediator role of platelet-derived growth factor and ERK. J Biol Chem. 2002;277:23165–71. doi: 10.1074/jbc.M201409200. [DOI] [PubMed] [Google Scholar]
- 13.Morrow D, Sweeney C, Birney YA, Cummins PM, Walls D, Redmond EM, Cahill PA. Cyclic strain inhibits Notch receptor signaling in vascular smooth muscle cells in vitro. Circ Res. 2005;96:567–575. doi: 10.1161/01.RES.0000159182.98874.43. [DOI] [PubMed] [Google Scholar]
- 14.Morrow D, Scheller A, Birney YA, Sweeney C, Guha S, Cummins PM, Murphy R, Walls D, Redmond EM, Cahill PA. Notch-mediated CBF-1/RBP-J{kappa}-dependent regulation of human vascular smooth muscle cell phenotype in vitro. Am J Physiol Cell Physiol. 2005;289:C1188–1196. doi: 10.1152/ajpcell.00198.2005. [DOI] [PubMed] [Google Scholar]
- 15.Proweller A, Pear WS, Parmacek MS. Notch signaling represses myocardin-induced smooth muscle cell differentiation. J Biol Chem. 2005;280:8994–9004. doi: 10.1074/jbc.M413316200. [DOI] [PubMed] [Google Scholar]
- 16.Tang Y, Urs S, Liaw L. Hairy-related transcription factors inhibit Notch-induced smooth muscle alpha-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circ Res. 2008;102:661–8. doi: 10.1161/CIRCRESAHA.107.165134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sweeney C, Morrow D, Birney YA, Coyle S, Hennessy C, Scheller A, Cummins PM, Walls D, Redmond EM, Cahill PA. Notch 1 and 3 receptor signaling modulates vascular smooth muscle cell growth. apoptosis, and migration via a CBF-1/RBP-Jk dependent pathway. FASEB J. 2004;18:1421–3. doi: 10.1096/fj.04-1700fje. [DOI] [PubMed] [Google Scholar]
- 18.Zhu GM, Guo HE, Zhang WW. Knockdown of Notch3 expression in VSMC by RNA interference. Chinese Journal of Geriatric Heart Brain and Vessel Diseases. 2011;12:1127–9. [Google Scholar]
- 19.Ishiko A, Shimizu A, Nagata E, Takahashi K, Tabira T, Suzuki N. Notch3 ectodomain is a major component of granular osmiophilic material (GOM) in CADASIL. Acta Neuropathologica. 2006;112:333–339. doi: 10.1007/s00401-006-0116-2. [DOI] [PubMed] [Google Scholar]
- 20.Boucher J, Gridley T, Liaw L. Molecular pathways of notch signaling in vascular smooth muscle cells. Front Physiol. 2012;3:81. doi: 10.3389/fphys.2012.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monet-Leprêtre M, Bardot B, Lemaire B, Domenga V, Godin O, Dichgans M, Tournier-Lasserve E, Cohen-Tannoudji M, Chabriat H, Joutel A. Distinct phenotypic and functional features of CADASIL mutations in the Notch3 ligand binding domain. Brain. 2009;132:1601–1612. doi: 10.1093/brain/awp049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D’Souza B, Meloty-Kapella L, Weinmaster G. Canonical and non-canonical Notch ligands. Curr Top Dev Biol. 2010;92:73–129. doi: 10.1016/S0070-2153(10)92003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fortini ME. Notch signaling: the core pathway and its posttranslational regulation. Dev Cell. 2009;16:633–647. doi: 10.1016/j.devcel.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 24.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Yang G, Zhang X, Wu J, Gu Q, Wei M, Yang J, Zhu Y, Wang N, Guan Y. Induction of MIF expression by oxidized LDL via activation of NF-kappaB in vascular smooth muscle cells. Atherosclerosis. 2009;207:428–433. doi: 10.1016/j.atherosclerosis.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 26.Tharmalingam S, Daulat AM, Antflick JE, Ahmed SM, Nemeth EF, Angers S, Conigrave AD, Hampson DR. Calcium-sensing receptor modulates cell adhesion and migration via integrins. J Biol Chem. 2011;286:40922–33. doi: 10.1074/jbc.M111.265454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romer LH, Birukov KG, Garcia JG. Focal adhesions: paradigm for a signaling nexus. Circ Res. 2006;98:606–616. doi: 10.1161/01.RES.0000207408.31270.db. [DOI] [PubMed] [Google Scholar]
- 28.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. Schlaepfer, FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 29.Schober M, Raghavan S, Nikolova M, Polak L, Pasolli HA, Beggs HE, Reichardt LF, Fuchs E. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J Cell Biol. 2007;176:667–680. doi: 10.1083/jcb.200608010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song GJ, Leslie KL, Barrick S, Bougoin S, Taboas JM, Bisello A. EBP50 promotes focal adhesion turnover and vascular smooth muscle cells migration. J Mol Cell Cardiol. 2012;53:809–819. doi: 10.1016/j.yjmcc.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.