

Abstract
Neural stem cells (NSCs) are multi-potent stem cells able to self-renew and generate immature and differentiated cell populations by asymmetric division. The NSCs are of considerable interest for cell replacement in neuro-degenerative diseases. NSCs are usually identified and expanded by their ability to generate free-floating aggregates termed neurospheres. However, neurospheres are not a pure population of NSCs with as little as 1% population in primary spheres. Neurospheres also contain neurons, astrocytes and oligodendrocytes. The heterogeneity of these cells may hinder their repopulation potential when used in cell transplantation. Furthermore, to obtain 1 million NSCs by the neurosphere protocol usually takes one month, which is inconvenient for future clinical trials. In this study, we tried to derive the NSCs from mice embryo neuroepithelium without neurosphere formation. Three different protocols were compared. We generated a direct and efficient NSCs generation, expanding and freezing protocol. This protocol can provide sufficient amount of the NSCs from first a few passages for cell transplantation.
Keywords: Neural stem cell, neuro-sphere, differentiation, neuron
Introduction
The characteristics of neural stem cells (NSCs) are of considerable interest, not only for understanding their roles in normal development of central nervous system, but also for cell replacement in neuro-degenerative diseases [1]. NSCs are multi-potent stem cells able to self-renew and generate immature and differentiated cell populations by asymmetric division [2]. NSCs resides initially in the early neuroepithelium and later in the ventrical zone (VZ) and subventricular zone (SVZ) during embryogenesis, and they exists in some areas of the adult brain [3]. NSCs markers, such as intermediate filament protein Nestin [4], Prominutesin/CD133 [5,6], and CD15 [7] are currently used to identify stem cells in the nervous system. These markers are selective rather than specific for NSCs. NSCs have been shown to have high telomerase [8] and aldehyde dehydrogenase activity [9,10].
Two strategies are being considered for adult NSC-based therapy, the stimulation of endogenous neural progenitor cells and the transplantation of adult-derived NSCs. The stimulation of endogenous NSCs would generate in the SVZ and migrate to the sites of degeneration in the Huntington’s disease and stroke [11-13]. Adult derived-neural progenitor cells were administrated intravenously to promote repair [14]. Systemic injection provides a model of delivering NSCs for the treatment of neurological disease with widespread degeneration, such as Huntington’s disease and Alzheimer’s disease.
NSCs are usually identified and expanded by their ability to generate free-floating aggregates termed neurospheres [15]. However, neurospheres are not a pure population of NSCs with as little as 1% population in primary spheres. Neurospheres also contain neurons, astrocytes and oligodendrocytes [16-19]. The heterogeneity of these cells may hinder their repopulation potential when used in cell transplantation. The neurosphere generation protocol cannot provide primary NSCs efficiently. In this study, we tried to derive NSCs from mice embryo neuroepithelium without neurosphere formation. Three different protocols were compared. We generated a direct and efficient NSCs generation, expanding and freezing protocol. This protocol can provide sufficient amount of NSCs from first a few passages for cell transplantation.
Materials and methods
Comparison of three mouse embryo NSC generation protocols
The 6 well plates were coated with 20 mg/ml Poly-L-ornithine (Sigma, P4957) for at least two hours in the incubator. The Poly-L-ornithine solution was taken away, and the plate was rinsed once with water. 5 mg/ml Laminine (Sigma, L2020) was added into the plate and stayed in the incubator for one hour. Use PBS water to rinse once just before use.
SCR pregnant mice were housing according to the Tongji University Animal Affair Standards. On day 15 of pregnancy, pregnant mouse was sacrificed and mouse embryos were taken out. The outer membrane of embryo brain was peeled off and neuroepithelium tissue were isolated and minced with scissor. HBSS solution (without Ca2+ and Mg2+) (Life Technologies, 14170112) with 20% fetal bovine serum (FBS) (Life Technologies, 10439001) was used to rinse the minced tissue twice. HBSS solution without Ca2+ and Mg2+ was used to rinse the minced tissue for further three times to get rid of blood cells. Equal amount of the minced tissue was separated into three 15 ml centrifuge tubes. Different digestion enzymes and incubation time were used to digest the minced tissue. They were: 0.25% Trypsin (Life Technologies, 15050057), 10 minutes; 0.25% Trypsin plus 15 ml Deoxyribonuclease 1 (Worthington, LS006353), 10 minutes; and Papain solution (Worthington, LS003118), 20 minutes. 3 ml of HBSS solution with 20% FBS was added into the mixture, centrifuged at 1500 RPM for 3 minutes, at 4°C. The supernatant was thrown away, the cell pellet was re-suspended with NSC grow culture medium, which contained 87% of DMEM/F12 (Life Technologies, 11039021), 10% FBS, 1% L-Glutamine (Life Technologies, 25030149), 2% B27 (Life Technologies, 17504044). The cell suspension was filtered through 70 micron pores nylon mesh (BD Falcon, 352350), and plated onto the coated 6-well plates. On the first day of cell culture, the primary NSCs were cultured with NSC growth medium with 10% FBS, 40 ng/ml FGF2, 20 ng/ml EGF and 20 ng/ml PDGF. On the second day, the primary NSCs were cultured with NSC growth medium without FBS, with 40 ng/ml FGF2, 20 ng/ml EGF and 20 ng/ml PDGF. After that, 50% of the culture medium was changed with fresh NSC growth medium, add fresh growth factors. Store the used NSC growth medium at 4°C.
Comparison of two primary NSC passage protocols
Prepare HBS solution (HBSS solution without Ca2+ and Mg2+, 350 mg/L NaHCO3, 1 mM Hepes, adjust PH to 7.3-7.4). When the primary NSCs became confluent on the 6-well plates, get rid of the growth medium, rinse once with HBSS solution. Add 1 ml chemical digestion solution (5 ml HBS solution, 12 mM MgSO4) or 0.25% Trypsin solution, 37°C, 5 minutes. Add 7 ml HBSS solution with 20% FBS, centrifuge at 1500RPM for 3 minutes. Discard the supernatant, re-suspend the cell pellet with NSC growth medium, plate on the coated 6-well plates, and expand at 1:3 or 1:6 ratios.
Cryopreservation of the NSCs
Collect the NSCs with 0.25% Trypsin, centrifuge at 1500RPM for 3 minutes, and re-suspend the cell pellet with 90% of the collected used the NSC growth medium and 10% DMSO. 1 well of the NSCs on the 6-well plate shall be stored into 1 vial of freeze tube. Samples were gradually cooled to -80°C in isopropanol freezing containers. Frozen samples were then transferred to liquid nitrogen for long-term storage. When thaw the frozen vial of NSCs, take the vial out of liquid nitrogen, thaw in 37°C water bath for 5 minutes. Add 5 ml HBSS solution the NSC growth medium with 20% FBS, centrifuge at 1500RPM for 3 minutes. Re-suspend the cell pellet with NSC growth medium with 10% FBS, 40 ng/ml FGF2, 20 ng/ml EGF and 20 ng/ml PDGF. Plate the NSCs on coated 6-well plate. 1 vial of frozen NSC shall be plated on one well of 6-well plate.
Characterization of the mouse embryo NSCs by polymerase chain reaction
Total RNA was extracted using RNeasy™ columns (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. Total RNA (1 µg) was reverse transcribed into cDNA using the NCode™ miRNA First-Strand cDNA Synthesis Kit (Clontech Laboratories Inc., Mountain View, CA, USA). The primer sequences for characterizing the NSCs were: EGFR forward primer (acactgctggtgttgctgac), reverse primer (cccaaggaccacttcacagt); Sox2 forward primer (cacaactcggagatcagcaa), reverse primer (ctccgggaagcgtgtactta); Pax6 forward primer (agggggagagaacaccaact), reverse primer (tttggcccttcgattaga); Nestin forward primer (aggctgagaactctcgcttg), reverse primer (attaggcaagggggaagaga); GFAP forward primer (cacgaacgagtccctagagc), reverse primer (atggtgatgcggttttcttc). The cycling programme involved preliminary denaturation at 95°C for 10 minutes, followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 56°C for 30 s, and elongation at 72°C for 30 s, followed by a final elongation step at 72°C for 10 minutes.
Characterization of the mouse embryo NSCs by immunohistochemistry
The cells were fixed with 4% paraformaldehyde for 10 minutes at room temperature. After being blocked with 5% normal goat or donkey serum in PBS, cells were incubated with primary antibody overnight at 4°C. The following proteins were evaluated: Nestin (mouse monoclonal, 1:200, Chemicon, 2C13B9); Sox1 (rabbit polyclonal, 1:200, Chemicon, AB5768); b-III-tubulin (TuJ-1, mouse monoclonal, 1:200, Chemicon, MAB1637); glial fibrillary acid protein (GFAP, 1:400, Sigma, SAB1405864); Synapsin (1:500, Sigma, SAB4502905). After being washed for 3 times with PBS, the cells were incubated with secondary antibody for 2 hours at room temperature. The following secondary antibodies were used: FITC-AffiniPure Goat anti-Mouse IgG (Jackson ImmunoResearch, 115-095-205), Alexa Fluro®647-AffiniPure Goat anti-Mouse IgG (Jackson ImmunoResearch, 115-605-205), Alexa Fluro®647-AffiniPure F (ab’) 2 Fragment Donkey anti-Rabbit IgG (H+L) (Jackson ImmunoResearch, 711-606-152). The labeled slides were dehydrated in alcohol and xylene before being cover-slipped with mounting media.
Glial Culture
The minced neuroepithelium tissue was plated on non-coated 6-well plates, cultured with DMEM medium (Life Technologies, 10569044) with 1% L-Glutamine, 10% FBS and 1% Penicillin-Streptomycin (Life Technologies, 10378016). The culture media was changed once per week at the beginning. When there were more glial cells, the culture media was changed more frequently. When the glial cells were nearly confluent, the culture media was changed into neurobasal medium (Life Technologies, 0050128DJ) with B27 (Life Technologies, 0080085-SA). The next day, collect the media and change into DMEM medium, with 1% L-Glutamine, 10% FBS and 1% Penicillin-Streptomycin. After 24 hours, discard the media and change into neurobasal medium with B27, which was collected after 24 hours. These two different culture media was used in rotation. The media could be collected in one week culture of confluent glial cells.
NSC differentiation into neurons
Mix the collected glial cell culture medium with DMEM and L-Glutamine at the ratio of 1:1, plus 1% FBS, 100 nM all-trans-retinoic acid (Sigma, R2500), 20 ng/ml brain derived neurotrophic factor (BDNF, Millipore, 203702), 20 ng/ml neurotrophin 3 (NT-3, Millipore, GF308).
Results
Primary NSCs derivative efficiency comparison
The mouse fetus neuroepithelium tissue was minced and digested with three different ways, 0.25% Trypsin for 10 minutes, 0.25% Trypsin plus 15 ml Deoxyribonuclease for 10 minutes, and Papain solution for 20 minutes. The primary NSCs were cultured with NSC growth medium with 10% FBS, 40 ng/ml FGF2, 20 ng/ml EGF and 20 ng/ml PDGF. On the second day, the primary NSCs were cultured with NSC growth medium without FBS, with 40 ng/ml FGF2, 20 ng/ml EGF and 20 ng/ml PDGF. The images of the primary NSCs were taken on the second day of culture as shown in Figure 1. The primary NSCs derived using 0.25% Trypsin digestion method could provide the most amount of cells comparing with the other two digestion methods.
Figure 1.
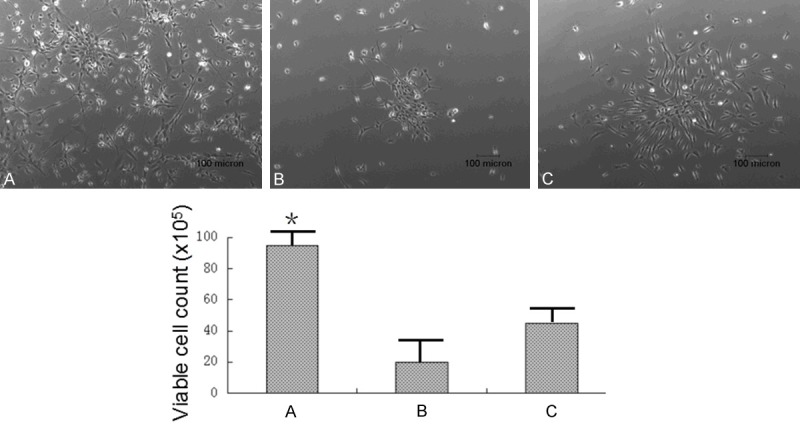
Primary NSCs derivative efficiency comparison. The mouse fetus neuroepithelium tissue was minced and digested with 0.25% Trypsin, 10 minutes (A); 0.25% Trypsin plus 15 µl Deoxyribonuclease, 10 minutes (B); and Papain solution, 20 minutes (C). The images of the primary NSCs were taken on the second day of culture. 0.25% Trypsin could derive NSCs more efficiently than the other digestion methods.
NSCs passage methods comparison
When the primary NSCs became confluent, the cells were digested with chemical digestion solution (5 ml HBS solution, 12 mM MgSO4) or 0.25% trypsin solution, 37°C for 5 minutes. The NSCs were expanded at 1:6 ratio. As shown in Figure 2, there were significant more NSCs attached to the plate bottom by using trypsin solution than using the chemical digestion method. 0.25% trypsin can passage the NSCs more efficiently than the chemical digestion method.
Figure 2.
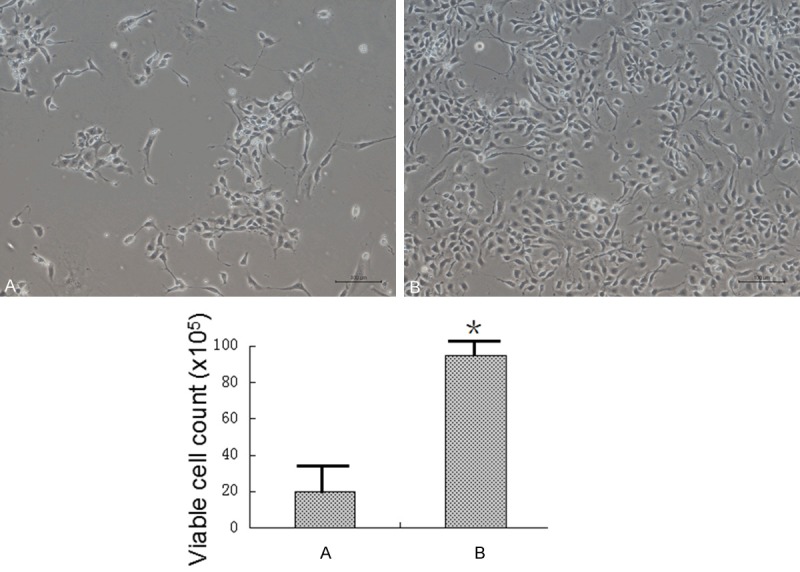
The NSCs passage methods comparison. When the primary NSCs became confluent, the cells were digested with Chemical digestion solution (A) or 0.25% Trypsin solution (B). The NSCs were expanded at 1:6 ratio. There were much more NSCs attached to the plate bottom by using Trypsin solution than using the Chemical digestion method (the scale bar stands for 100 μm).
Characterization of NSCs
The confluent primary NSCs were stained with SOX1 and Nestin antibodies. As shown in Figure 3, over 90% of the primary NSCs were SOX1 and Nestin positive. The initial three passages of the NSCs were harvested and RT-PCR was used to detect the known NSC markers. The results showed that the NSCs highly expressed Pax6, SOX2, Nestin, Epidermal growth factor receptor (EGFR) and Glial fibrillary acidic protein (GFAP).
Figure 3.
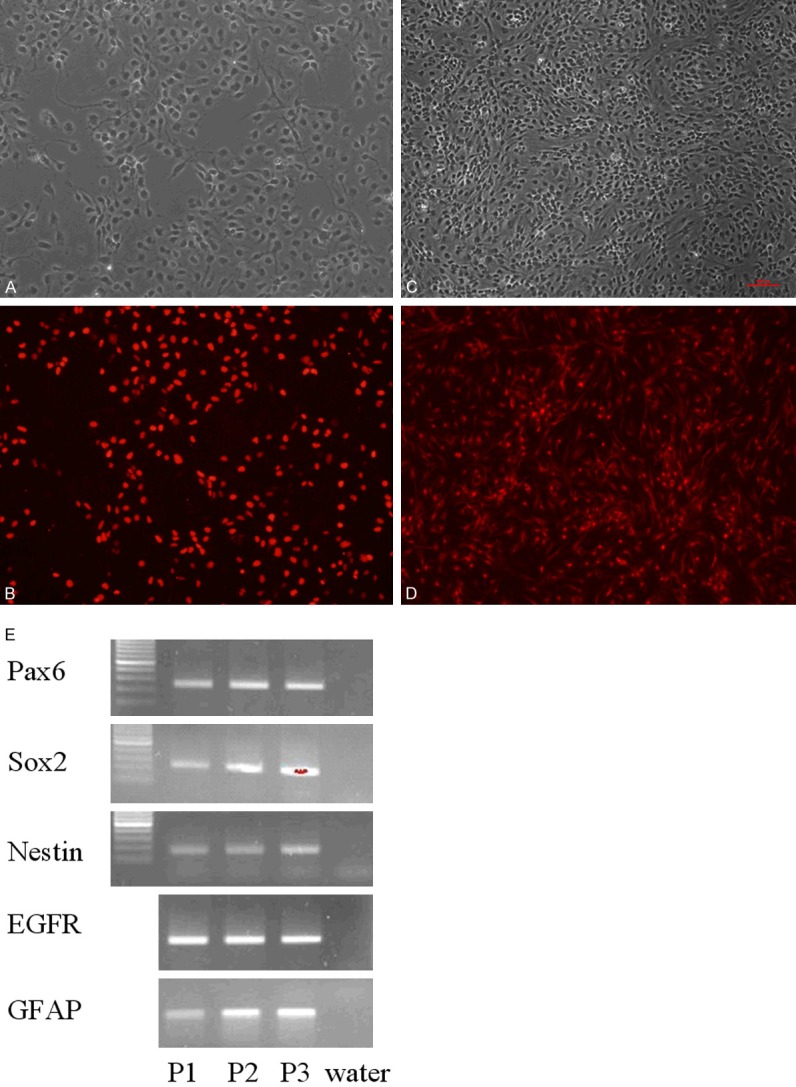
NSCs characterization. The NSC was stained with SOX1 (B) and Nestin antibodies (D). (A and C) are unstained corresponding images of (B and D). Over 90% of the NSCs were stained positive. The first three passage NSCs were harvested and intrinsic NSC markers, Pax6, Sox2, Nestin, EGFR and GFAP were tested by RT-PCR. Water was used as no loading control. The NSCs keep their NSC characteristic gene expression during passages.
NSCs differentiation into neurons
The NSCs were cultured with glial cell medium (collected glial cell culture medium) with all-trans-retinoic acid, BDNF and NT-3. After three weeks, the differentiated NSCs were stained with the Tuj1 antibody. Many neurons appeared as shown in Figure 4A. After 6 weeks, synapsins were formed clearly as shown in Figure 4B.
Figure 4.
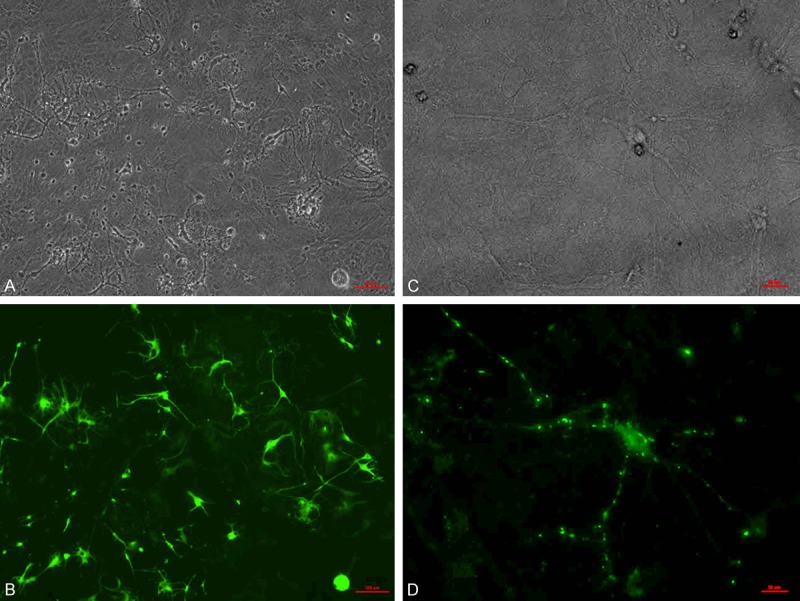
NSCs differentiation into neurons. The NSCs were cultured with glial cell medium with all-trans-retinoic acid, BDNF and NT-3. After three weeks, the differentiated NSCs were stained with the Tuj1 antibody. Many neurons appeared as shown in (A, B). After 6 weeks, synapsins were formed clearly as shown in (C, D). The red bar stands for 50 μm.
Discussion
Neurosphere method is commonly used to collect and expand the NSCs. In one primary neurosphere cell population, 99% cells are not the NSCs [16-19]. It generally believed that those non-NSCs provides proper growth niche for the NSCs. Without non-NSCs surround, the NSCs would not keep their stem cell characters, therefore the heterogeneity is necessary for the NSC growth.
One purpose of this paper is to test whether the NSCs could grow individually. By using 0.25% Trypsin to digest brain tissue, the primary NSCs were attached to the Poly-L-ornithine and laminine coated plates, and grown in the NSC growth medium containing the FGF2, EGF and PDGF. Over 95% of the attached cells were the NSCs by immunohistochemistry. These NSCs can be split into 1:6 during passage and grow confluent within three days. These NSCs also can be induced differentiated into neurons and form synapse. We conclude that the NSCs can grow individually without neurosphere.
Another purpose of this study is to develope a new protocol to provide the NSCs quicky and efficiently for future clinical trials. For collecting 1 million NSCs, it usually takes about one month using the neurosphere method. It often the case that the NSCs were contaminated before usage. By using this new protocol in this paper, it only takes about 10 days to collect over 1 million NSCs, and decreases the chances of contamination.
Conclusion
This paper provides a new method to collect and grow NSCs from the mice fetus brain tissue. Whether it can be used in the mice pap forbrain tissue needs to be studied further. Cell replacement research is needed to provide more research information by using the NSCs grown by this method.
Acknowledgements
Dr. Zhi-Zhao Ma did most lab works; Mr. Lin Fan and Dr. Jun-Ling Huang did the immunohistochemistry work; Dr. Xiao-Jing Pan organized results and wrote this paper.
Disclosure of conflict of interest
None.
References
- 1.Bjorklund A, Lindvall O. Cell replacement therapies for central nervous system disorders. Nat Neurosci. 2000;3:537–544. doi: 10.1038/75705. [DOI] [PubMed] [Google Scholar]
- 2.Wagers AJ, Weissman IL. Plasticity of adult stem cells. Cell. 2014;116:639–648. doi: 10.1016/s0092-8674(04)00208-9. [DOI] [PubMed] [Google Scholar]
- 3.Temple S. The development of neural stem cells. Nature. 2001;414:112–117. doi: 10.1038/35102174. [DOI] [PubMed] [Google Scholar]
- 4.Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 5.Corbeil D, Roper K, Hellwig A, Tavian M, Miraglia S, Watt SM, Simmons PJ, Peault B, Buck DW, Huttner WB. The human AC133 hematopoietic stem cell antigen is also expressed in epithelial cells and targeted to plasma membrane protrusions. J Biol Chem. 2000;275:5512–5520. doi: 10.1074/jbc.275.8.5512. [DOI] [PubMed] [Google Scholar]
- 6.Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capela A, Temple S. LeX/ssea-1 is expressed by adult mouse CNS stem cells, identifying them as nonependymal. Neuron. 2002;35:865–875. doi: 10.1016/s0896-6273(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 8.Cai J, Cheng A, Luo Y, Lu C, Mattson MP, Rao MS, Furukawa K. Membrane properties of rat embryonic multipotent neural stem cells. J Neurochem. 2004;88:212–226. doi: 10.1046/j.1471-4159.2003.02184.x. [DOI] [PubMed] [Google Scholar]
- 9.Corti S, Locatelli F, Papadimitriou D, Donadoni C, Salani S, Del Bo R, Strazzer S, Bresolin N, Comi GP. Identification of a primitive brain-derived neural stem cell population based on aldehyde dehydrogenase activity. Stem Cells. 2006;24:975–985. doi: 10.1634/stemcells.2005-0217. [DOI] [PubMed] [Google Scholar]
- 10.Storms RW, Trujillo AP, Springer JB, Shah L, Colvin OM, Ludeman SM, Smith C. Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci U S A. 1999;96:9118–9123. doi: 10.1073/pnas.96.16.9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taupin P. Adult neurogenesis, neuroinflammation and therapeutic potential of adult neural stem cells. Int J Med Sci. 2008;5:127–132. doi: 10.7150/ijms.5.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Curtis MA, Penney EB, Pearson AG, van Roon-Mom WM, Butterworth NJ, Dragunow M, Connor B, Faull RL. Increased cell proliferation and neurogenesis in the adult human Huntington’s disease brain. Proc Natl Acad Sci U S A. 2003;100:9023–9027. doi: 10.1073/pnas.1532244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- 14.Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, Dina G, Galli R, Del Carro U, Amadio S, Bergami A, Furlan R, Comi G, Vescovi AL, Martino G. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422:688–694. doi: 10.1038/nature01552. [DOI] [PubMed] [Google Scholar]
- 15.Temple S. Division and differentiation of isolated CNS blast cells in microculture. Nature. 1989;340:471–473. doi: 10.1038/340471a0. [DOI] [PubMed] [Google Scholar]
- 16.Reynolds BA, Weiss S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol. 1996;175:1–13. doi: 10.1006/dbio.1996.0090. [DOI] [PubMed] [Google Scholar]
- 17.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 18.Davis AA, Temple S. A self-renewing multipotential stem cell in embryonic rat cerebral cortex. Nature. 1994;372:263–266. doi: 10.1038/372263a0. [DOI] [PubMed] [Google Scholar]
- 19.Rietze RL, Valcanis H, Brooker GF, Thomas T, Voss AK, Bartlett PF. Purification of a pluripotent neural stem cell from the adult mouse brain. Nature. 2001;412:736–739. doi: 10.1038/35089085. [DOI] [PubMed] [Google Scholar]