

Abstract
Massive accumulation of amyloid beta (Aβ) has been implicated as a pivotal event in the pathogenesis of Alzheimer’s disease. The underlying mechanisms of Aβ-induced neurotoxicity include generation of reactive oxidative species (ROS), inflammation, and neurons loss. Allopregnano-lone (APα), a neurosteroid derive from neuroactive progesterone, has been demonstrated to have neuroprotective properties in vivo and vitro. In the present study, the effects of APα on oxidative damage in Aβ25-35-treated pheochromocytoma (PC12) cells were investigated. Pretreatment of APα significantly attenuated Aβ25-35-induced neuronal death. APα decreased the intracellular ROS generation and reduced lipid peroxidation induced by Aβ25-35. In addition, APα treatment enhanced antioxidant enzyme superoxide dismutase (SOD) activity. This study demonstrates that APα exerts a protective effect against Aβ25-35-induced neurotoxicity in PC12 cells. The protective role of APα likely results from inhibition of oxidative stress.
Keywords: Allopregnanolone, amyloid beta, reactive oxidative species, PC12 cells
Introduction
Alzheimer’s disease (AD), the most common neurodegenerative disorder, is characterized by the progressive deterioration of memory and cognition. The major neuropathological hallmarks of AD brain include intracellular senile plaques, neurofibrillary tangles, extensive neuronal death, and extracellular deposition of β-amyloid (Aβ) [1,2]. Aβ has been reported to be neurotoxic and plays a key role in the pathological cascade of AD [3]. Oxidative stress has been implicated in the pathological of AD and the excessive production of Aβ may trigger neuronal death by inducing free radical generation [4-6].
Allopregnanolone (APα), a neurosteroid synthesized from progesterone, appears to exert a pronounced neuroprotective effect in the setting of AD [7,8]. Recently, a reduction of APα level is found in serum [9], prefrontal cortex [10], and temporal cortex [11] of AD patients. The mechanism by which APα provide neuroprotection is not clearly understood. Previous findings show that APα increases proliferation of neural progenitor cells in vitro [12], promotes neurogenesis in the hippocampal [13], restores neural progenitor cell survival and cognitive performance in mouse model of AD [14].
Recent data indicate that APα has protective effect against several pathological conditions by restoring the intracellular redox status [15-17]. However, it has not been well elucidated whether APα exerts neuroprotection by reducing oxidative stress in AD. Therefore, we used the β-amyloid peptide Aβ25-35-treated pheochromocytoma (PC12) cells model to investigate the neuron-protective effect of APα. We aimed to investigate whether APα has protective effect against Aβ-induced neurotoxicity in PC12 cells and whether APα exerts this effect by reducing the intracellular oxidative stress.
Materials and methods
Drugs and reagents
Allopregnanolone (APα; 3α-hydroxy-5α-pregnan-20-one; aka AP, Allo, or THP), 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 2’,7’-dichlorofluorescin diacetate (DCFH-DA), β-amyloid peptide (Aβ25-35) and dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). RPMI 1640 medium and fetal bovine serum (FBS) were obtained from Gibco (Grand Island, NY, U.S.A.). Assay kits for superoxide dismutase (SOD) and malondialdehyde (MDA) were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). All other chemicals and reagents used were of analytical grade.
Drugs preparation
Aβ25-35 was dissolved in deionized distilled water at a concentration of 1 mM and incubated for 4 days at 37°C for aggregation. The solution was then stored at -20°C until use. APα was dissolved in DMSO at a concentration of 10 mM and further diluted with phosphate buffered saline (PBS). The final concentration of DMSO was 0.01%, which did not affect cell viability.
Cell culture
Pheochromocytoma (PC12) cells, a rat PC12 cell line, were obtained from the cell bank of Institute of Biochemistry and Cell Biology, SIBS, CAS (Shanghai, China). PC12 Cells were seeded in 25 cm2 flasks and maintained in RPMI 1640 supplemented with penicillin (100 U/mL), streptomycin (100 U/mL), and 5% FBS at 37°C in a humidified 95% air and 5% CO2 incubator. Once grown until 80% confluence, the cells were subcultured at an appropriate density according to each experimental scale at 37°C for 24 h in serum-free RPMI 1640. Then, the cells were used for treatment.
Cell viability assay
Cell viability following treatment with Aβ25-35, APα, or both was assessed by MTT assay. In brief, PC12 cells were seeded in 96 well plates at a density of 5 × 103 cells/well at 37°C for 24 h. After drug treatment, 20 μL MTT reagent (final concentration, 2.5 mg/mL) was added and the plate was incubated at 37°C for 4 h. The medium was removed and 150 μl DMSO was added. The absorbance of each well was read at 570 nm using a microplate reader (ELX808, Biotek, Winooski, VT, USA). Cell viability was expressed as a percentage of the value against the untreated control.
Measurement of malondialdehyde (MDA) and superoxide dismutase (SOD) content
For assay of lipid peroxide and antioxidants, cultured PC12 cells were initially seeded in 6 well plates at a density of 4 × 105 cells/ml for 24 h. The cells was then pre-incubated with or without APα (10 μM), followed by incubation with Aβ25-35 (20 μM) for 24 h. The cultures were washed with ice-cold PBS and homogenized. The homogenate was centrifuged at 12,000 × g for 15 min at 4°C. The protein concentration in each sample was determined by the BCA Protein Assay Kit as a reference standard. The levels of MDA and SOD were determined according to the manufacture’s instructions. Concentrations were normalized to the protein concentration expressed as a percentage of control samples.
Measurement of cellular generation of reactive oxygen species
Intracellular reactive oxygen species (ROS) was measured by using the fluorescence probe 2’, 7’-dichlorofluorescin diacetate (DCFH-DA). In brief, following the indicated drugs treatment, cells were washed twice with PBS, incubated with 10 μM DCFH-DA for 30 min at 37°C under the dark condition. After washed twice with PBS to remove the extracellular DCFH-DA, fluorescence was monitored at excitation and emission wavelengths of 485 and 530 nm, respectively. The fluorescence emission from DCF was analyzed via BD FACS Calibur flow cytometry (BD Science, San Jose, CA). Three independent samples of 10,000 cells were analyzed for each experimental condition. The mean fluorescence intensity was analyzed using FCS Express data analysis software.
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Biostatistical analyses were conducted with SPSS 16.0 software. Statistical differences among groups were assessed by one-way analysis of variance (ANOVA). Differences were considered to be statistically significant at P < 0.05.
Results
APα protects PC12 cells against Aβ25-35-induced cell injury
The Aβ25-35-induced cell injury was determined by the percentage of MTT reduction assay. As shown in Figure 1A, Aβ25-35 (2.5, 5, 10, 20, 40 μM) induced cell death in a dose-dependent manner. Nearly only 50% of PC12 cell survived after 24 h treatment with 20 μM of Aβ25-35. Based on the result, 20 μM was selected as the optimal Aβ25-35 concentration for subsequent experiments. APα at 10 μM showed strong inhibitory activity against Aβ25-35 induced cell death (Figure 1B). Furthermore, protective role of APα against Aβ25-35 induced cell death was confirmed by morphologic changes (Figure 1C). PC12 cells exposed to Aβ25-35 (20 μM) for 24 h led to cell nucleus pycnosis, nuclear fragmentation, and cell number reduction. Pretreatment with 10 μM APα for 2 h maintained normal cell morphology and cell number.
Figure 1.
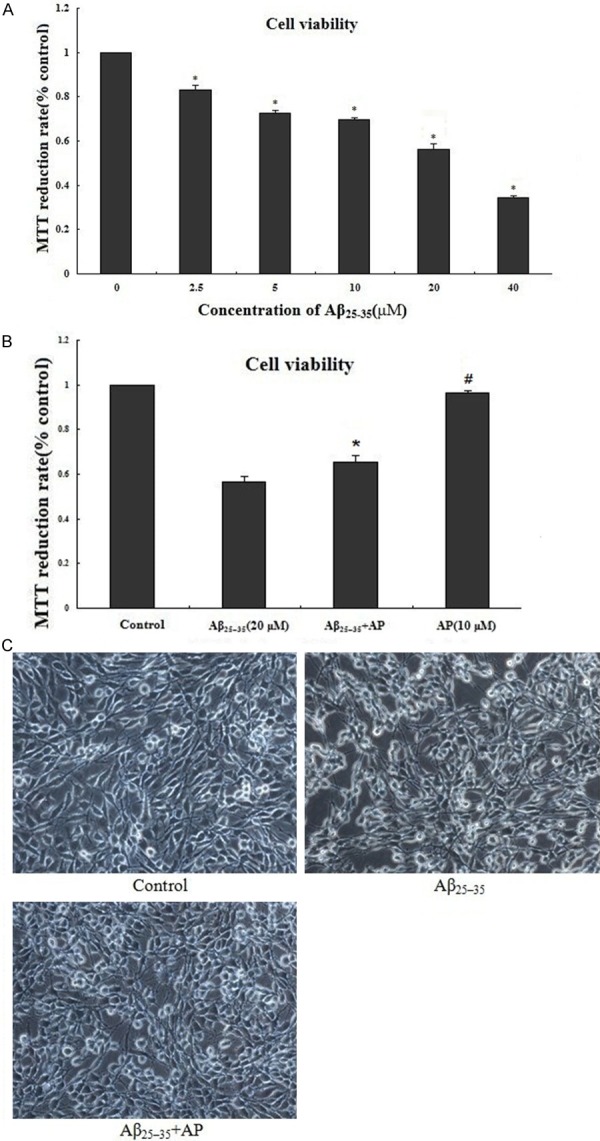
Protective effect of APα on Aβ25-35-induced cytotoxicity in cultured differentiated PC12 cells. A: PC12 cells were treated with the indicated concentrations (0-40 μM) of Aβ25-35 for 24 h; B: PC12 cells were pretreated with different concentrations of APα (10 μM) for 2 h and then incubated with Aβ25-35 (20 μM) for an additional 24 h. Viability of cells was assessed by MTT assay; C: Representative photographs of cell morphology by living cell picture (400 ×). PC12 cells treated with only Aβ25-35 can lead to cell nucleus pycnosis, nuclear fragmentation and reduce the number of cells. Application of 10 μM APα can maintain normal cell morphology and cell number. Percentage of cell viability was relative to the untreated control cells. Values represent as mean ± SD of 3 independent experiments. *P < 0.01 versus Aβ25-35-treated cells; #P > 0.05 versus control.
APα attenuated oxidative stress in Aβ25-35-treated PC12 cells
Oxidative stress induced by Aβ25-35 was determined by measurement of intracellular ROS (Figure 2). After treatment of PC12 cells with 20 μM Aβ25-35 for 24 h, intracellular ROS level increased to 187.31 ± 29.41. Thus, Aβ25-35 markedly increased oxidative stress. Pretreatment with 10 μM of APα for 2 h, intracellular ROS level induced by Aβ25-35 decreased to 147.78 ± 31.44.
Figure 2.
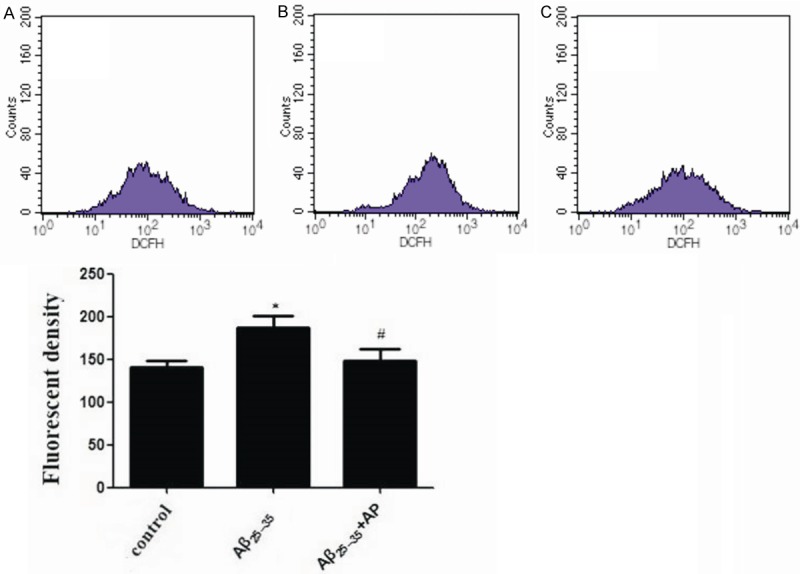
Effects of Aβ25-35 and APα on intracellular ROS in PC12 cells. Cells were treated with: A: Control (untreated); B: 20 μM Aβ25-35; C: 20 μM Aβ25-35 + 10 μM APα. Intracellular ROS were measured by flow cytometry using an oxidation-sensitive fluorescent probe, DCFH-DA in the presence of ROS.
APα attenuated lipid peroxidation in Aβ25-35-treated PC12 cells
To further confirm APα attenuates oxidative damage in Aβ25-35-treated PC12 cells, we measured the intracellular level of MDA, a marker of lipid peroxidation (Table 1). After treatment of PC12 cells with 20 μM Aβ25-35 for 24 h, intracellular MDA levels were 305.1% of control. While with the pretreatment of 10 μM of APα for 2 h, intracellular MDA level induced by Aβ25-35 decreased to 202.6% of control.
Table 1.
Effects of APα on lipid peroxidation and antioxidantenzyme activities in Aβ25-35-treated cultured PC-12 cells
| Treatment | MDA (nM/mg protein) | SOD (U/mg protein) |
|---|---|---|
| Control | 0.78 ± 0.23 | 50.2 ± 9.9 |
| Aβ25-35 | 3.16 ± 1.00* | 32.2 ± 9.4* |
| AP + Aβ25-35 | 1.58 ± 0.67# | 45.4 ± 8.6# |
Cells were pretreated with 10 μM APα for 2 h and then incubated with Aβ25-35 (20 μM) for an additional 24 h. Data were presented as mean ± SD.
P < 0.05 versus control;
P < 0.05 versus Aβ25-35-treated cells.
APα rescued loss of antioxidant enzyme activities in Aβ25-35-treated PC12 cells
To investigate the mechanisms may involve in the protective effects of APα, the level of antioxidant enzyme SOD (Table 1) were measured. After treatment of PC12 cells with 20 μM Aβ25-35 for 24 h, intracellular SOD expression level reduced to 64.1% of control. Thus, Aβ25-35 markedly reduced antioxidant capacity. 10 μM APα treatment for 2 h enhanced SOD activity to 90.4% of control.
Discussion
In the current study, we showed that APα exerted protective effect against Aβ25-35-induced neurotoxicity in PC12 cells. The protective role of APα was through inhibition of intracellular oxidative stress.
Our data suggest that treatment of PC12 cells with Aβ25-35 induced obvious neurotoxicity as indicated by enhanced cell death. Treatment with APα significantly decreased Aβ25-35 induced cell death, suggesting the protective effects of APα against Aβ25-35-induced neurotoxicity.
Since substantial evidence showed the important role of oxidative stress in the pathophysiology of AD [18,19] and Aβ exerts neurotoxicity by generating ROS [4-6,20,21]. We next to investigate the effect of APα on intracellular ROS production induced by Aβ25-35. Our results show that treatment of PC12 cells with Aβ25-35 markedly increased intracellular ROS production. Pretreatment with APα significantly inhibited intracellular ROS level induced by Aβ25-35. These results suggest that APα may acts as an antioxidant against Aβ25-35-induced neurotoxicity, which is consistent with the finding of previous research that APα has protective effect against several pathological conditions by reducing intracellular ROS production [15-17]. A previous study indicated that APα treatment significantly reduced the ROS accumulation and lipid peroxidation in human Niemann Pick C (NPC) fibroblasts, and prevented peroxide-induced apoptosis [15]. Another report had explored the contribution of APα in the protective effect of palmitoylethanolamide against oxidative stress, and found that the reduction of oxidative stress by palmitoylethanolamide was mediated through APα synthesis in astrocytes [16]. APα had also been shown to markedly prevent high glucose-induced oxidative damage in PC12 cells [17]. Our results also indicate that treatment of PC12 cells with Aβ25-35 markedly increased lipid peroxidation and APα treatment significantly inhibited this effect, which further confirmed APα acts as an antioxidant against Aβ25-35-induced neurotoxicity.
The mechanism by which APα restores the intracellular redox status is not clear. Zampieri et al. had shown that APα does not act as a free radical scavenger and APα might reduce the intracellular ROS production by increasing catalase activity [15]. In this previous study, the authors also suggested that APα might protect NPC cells from peroxide-induced cell death by suppressing NF-κB signaling [15]. Our data indicate that APα might reduce the intracellular concentrations of ROS by enhancing endogenous antioxidant enzyme SOD activity. The molecular mechanism by which APα restores SOD activity is not clear and needs to be further investigated. Whether APα protects PC12 cells from Aβ25-35-induced cell death by suppressing NF-κB signaling also needs to be further explored.
In conclusion, the present study suggests that APα exerts a protective effect against Aβ25-35-induced neurotoxicity in PC12 cells and APα could be a promising therapeutic strategy for treatment of AD. The protective role of APα at least in part results from inhibition of oxidative stress.
Disclosure of conflict of interest
None.
References
- 1.Selkoe DJ. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis. 2001;3:75–80. doi: 10.3233/jad-2001-3111. [DOI] [PubMed] [Google Scholar]
- 2.Katzman R, Saitoh T. Advances in Alzheimer’s disease. FASEB J. 1991;5:278–86. [PubMed] [Google Scholar]
- 3.Li MH, Jang JH, Sun B, Surh YJ. Protective effects of oligomers of grape seed polyphenols against beta-amyloid-induced oxidative cell death. Ann N Y Acad Sci. 2004;1030:317–29. doi: 10.1196/annals.1329.040. [DOI] [PubMed] [Google Scholar]
- 4.Muthaiyah B, Essa MM, Chauhan V, Chauhan A. Protective effects of walnut extract against amyloid beta peptide-induced cell death and oxidative stress in PC12 cells. Neurochem Res. 2011;36:2096–103. doi: 10.1007/s11064-011-0533-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen DL, Zhang P, Lin L, Shuai O, Zhang HM, Liu SH, Wang JY. Protective effect of Bajijiasu against β-amyloid-induced neurotoxicity in PC12 cells. Cell Mol Neurobiol. 2013;33:837–50. doi: 10.1007/s10571-013-9950-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hong SY, Jeong WS, Jun M. Protective effects of the key compounds isolated from Corni fructus against β-amyloid-induced neurotoxicity in PC12 cells. Molecules. 2012;17:10831–45. doi: 10.3390/molecules170910831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irwin RW, Solinsky CM, Brinton RD. Frontiers in the therapeutic development of the allopregnanolone for Alzheimer’s disease and other neurological disorders. Front Cell Neurosci. 2014;8:203. doi: 10.3389/fncel.2014.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irwin RW, Brinton RD. Allopregnanolone as regenerative therapeutic for Alzheimer’s disease: translational development and clinical promise. Prog Neurobiol. 2014;113:40–55. doi: 10.1016/j.pneurobio.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernardi F, Lanzone A, Cento RM, Spada RS, Pezzani I, Genazzani AD, Luisi S, Luisi M, Petraglia F, Genazzani AR. Allopregnanolone and dehydroepiandrosterone response to corticotropin-releasing factor in patients suffering from Alzheimer’s disease and vascular dementia. Eur J Endocrinol. 2000;142:466–71. doi: 10.1530/eje.0.1420466. [DOI] [PubMed] [Google Scholar]
- 10.Marx CE, Trost WT, Shampine LJ, Stevens RD, Hulette CM, Steffens DC, Ervin JF, Butterfield MI, Blazer DG, Massing MW, Lieberman JA. The neurosteroid allopregnanolone is reduced in prefrontal cortex in Alzheimer’s disease. Biol Psychiatry. 2006;60:1287–94. doi: 10.1016/j.biopsych.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 11.Naylor JC, Kilts JD, Hulette CM, Steffens DC, Blazer DG, Ervin JF, Strauss JL, Allen TB, Massing MW, Payne VM, Youssef NA, Shampine LJ, Marx CE. Allopregnanolone levels are reduced in temporal cortex in patients with Alzheimer’s disease compared to cognitively intact control subjects. Biochim Biophys Acta. 2010;1801:951–9. doi: 10.1016/j.bbalip.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang JM, Johnston PB, Ball BG, Brinton RD. The neurosteroid allopregnanolone promotes proliferation of rodent and human neural progenitor cells and regulates cell-cycle gene and protein expression. J Neurosci. 2005;25:4706–18. doi: 10.1523/JNEUROSCI.4520-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang JM, Singh C, Liu L, Irwin RW, Chen S, Chung EJ, Thompson RF, Brinton RD. Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2010;107:6498–503. doi: 10.1073/pnas.1001422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh C, Liu L, Wang JM, Irwin RW, Yao J, Chen S, Henry S, Thompson RF, Brinton RD. Allopregnanolone restores hippocampal-dependent learning and memory and neural progenitor survival in aging 3xTgAD and nonTg mice. Neurobiol Aging. 2012;33:1493–506. doi: 10.1016/j.neurobiolaging.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zampieri S, Mellon SH, Butters TD, Nevyjel M, Covey DF, Bembi B, Dardis A. Oxidative stress in NPC1 deficient cells: protective effect of allopregnanolone. J Cell Mol Med. 2009;13:3786–96. doi: 10.1111/j.1582-4934.2008.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raso GM, Esposito E, Vitiello S, Iacono A, Santoro A, D’Agostino G, Sasso O, Russo R, Piazza PV, Calignano A, Meli R. Palmitoylethanolamide stimulation induces allopregnanolone synthesis in C6 Cells and primary astrocytes: involvement of peroxisome-proliferator activated receptor-α. J Neuroendocrinol. 2011;23:591–600. doi: 10.1111/j.1365-2826.2011.02152.x. [DOI] [PubMed] [Google Scholar]
- 17.Afrazi S, Esmaeili-Mahani S, Sheibani V, Abbasnejad M. Neurosteroid allopregnanolone attenuates high glucose-induced apoptosis and prevents experimental diabetic neuropathic pain: in vitro and in vivo studies. J Steroid Biochem Mol Biol. 2014;139:98–103. doi: 10.1016/j.jsbmb.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 18.Culmsee C, Landshamer S. Molecular insights into mechanisms of the cell death program: role in the progression of neurodegenerative disorders. Curr Alzheimer Res. 2006;3:269–83. doi: 10.2174/156720506778249461. [DOI] [PubMed] [Google Scholar]
- 19.Brinton RD, Wang JM. Therapeutic potential of neurogenesis for prevention and recovery from Alzheimer’s disease: allopregnanolone as a proof of concept neurogenic agent. Curr Alzheimer Res. 2006;3:185–90. doi: 10.2174/156720506777632817. [DOI] [PubMed] [Google Scholar]
- 20.Kong JJ, Zhang DD, Li P, Wei CY, Yu HJ, Zhang H, Zhang W, Wang YF, Cao YP. Nicorandil inhibits oxidative stress and amyloid-β precursor protein processing in SH-SY5Y cells overexpressing APPsw. Int J Clin Exp Med. 2015;8:1966–75. [PMC free article] [PubMed] [Google Scholar]
- 21.Ye J, Zhang Y. Curcumin protects against intracellular amyloid toxicity in rat primary neurons. Int J Clin Exp Med. 2012;5:44–9. [PMC free article] [PubMed] [Google Scholar]