

Abstract
Biosimilars are now a reality in rheumatology. Although analytical and non-clinical procedures to establish similarity have evolved significantly, clinical trials demonstrating equivalent efficacy and safety are absolutely required for all biosimilars. The design of such trials, including equivalence and non-inferiority statistical approaches, are discussed. Clinical evidence on biosimilars that have been approved recently or are presently being developed for use in rheumatology is also reviewed and contrasted with that available for biomimics (or intended copies), which are non-innovator biologics that are marketed in several countries but have not undergone review according to a regulatory pathway for biosimilars.
Keywords: Outcomes research, Pharmacokinetics, Anti-TNF
Key messages.
What is already known about the subject?
Biosimilars are approved following a regulatory pathway different to that of generics, as they are not molecularly identical to their reference products. Thorough information must be provided on analytical and non-clinical procedures to show similarity. Moreover, clinical trials demonstrating equivalent efficacy and safety to the reference product are absolutely required for all biosimilars.
What does this study add?
The design of equivalence and non-inferiority clinical trials is described and discussed. Clinical evidence available for biosimilars is reviewed and contrasted to that of biomimics (or intended copies), which are non-innovator products being commercialized in some countries without undergoing review according to a regulatory pathway for biosimilars.
How might this impact in clinical practice?
Familiarity with issues regarding study design and methods of analysing data obtained in comparative clinical trials allows improving decision making surrounding the prescription of biosimilars to patients with rheumatic diseases.
Background
There is now considerable interest in biosimilars among rheumatologists, although the distinction between a true biosimilar and a biomimic (or intended copy) may not be clear to most. Many countries have changed their regulatory requirements to accommodate this new class of medicinal products and to distinguish them from generics.1 2 Currently, only one biosimilar is approved by the European Medicines Agency (EMA) for the treatment of rheumatological diseases: an infliximab biosimilar, which is commercialised as Remsima/Inflectra. Following the lead of the EMA, regulatory agencies in other countries, including South Korea, Canada, Japan, Turkey and Colombia,3 have approved this infliximab biosimilar. However, the approved indications differ among these countries. For example, EMA allowed the results of clinical trials conducted in rheumatological diseases trials to be extrapolated to inflammatory bowel diseases, while Health Canada did not.4 Recently, BOW015, an infliximab biosimilar with the commercial name Infimab,5 and ZRC-3197, an adalimumab biosimilar with the commercial name Exemptia,6 were approved in India, while HD203, an etanercept biosimilar, was approved in South Korea.7 Whether other countries will approve these products with the data currently available, however, remains to be seen.
Currently, numerous biosimilars are in development and it is likely that some of them will be commercialised in the near future.2 Although analytical and non-clinical procedures to establish similarity have evolved significantly, clinical trials demonstrating equivalent efficacy and safety are an absolute requirement for regulatory approval of all biosimilars.2 8 Understanding the principles by which these trials are designed and analysed will help the clinician to evaluate and use these drugs in practice.
Design of clinical trials for biosimilarity
Pharmacokinetic studies
Pharmacokinetic comparisons demonstrating equivalence of certain biosimilars with their corresponding innovators have been conducted in healthy volunteers and in patients with rheumatological diseases.9–11 Pharmacokinetic equivalence is necessary, but not sufficient, to demonstrate biosimilarity.
Biosimilars are not identical to innovators. Certain molecular differences, albeit minimal, can modify affinity for the target ligand without modifying pharmacokinetics.2 8 Hence, innovator and non-innovator products may exhibit differences in clinical efficacy and safety despite comparable pharmacokinetics. This is why, unlike generic small molecule drugs, bioequivalence of a biosimilar with its reference product cannot be established solely on pharmacokinetic grounds.8 11
Small molecule generics are molecularly identical to the innovator small molecule drugs, such that all pharmacodynamic properties of both drugs are exactly the same. The same is not true of biosimilars with innovator biopharmaceuticals.12 Furthermore, a biosimilar and its reference biopharmaceutical can also differ in terms of immunogenicity.8 Thus, at least one clinical study documenting equivalent efficacy and safety is absolutely required.8 12
Equivalence studies
The purpose of a clinical trial comparing a biosimilar to its innovator is to demonstrate equivalent efficacy and safety. Therefore, a suitable patient population must be chosen that is sensitive enough for differences in the measured end point, which might be due to dissimilarities between the products assayed, to be detected.13 14 The patient groups receiving the innovator and the biosimilar must be balanced in terms of demographics and clinical characteristics. The number of subjects studied should be sufficient to achieve statistical power. Finally, the primary end point and the study duration must be clinically relevant.13
Hypothesis tests are designed to demonstrate differences between two samples and not equivalence.15 In a comparison performed using a hypothesis test, such as the Student's t test or analysis of variance, the absence of a significant difference (ie, p>0.05) does not demonstrate equivalence. A sample is described by its mean value and its variability, which is frequently expressed as the SD. If two samples overlap, it is difficult to demonstrate a significant difference. Overlapping can be due to a narrow difference between means or to wide SDs. The SD (s) of a sample is calculated by equation 1, where x and  are the individual and mean values of the measured parameter, and n the number of subjects.
are the individual and mean values of the measured parameter, and n the number of subjects.
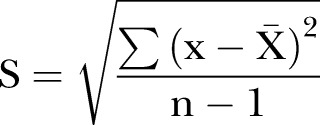 |
1 |
If the difference between mean values is the same, overlapping depends on the SD values of both samples. This is shown in figure 1, where two comparisons are performed where the difference between mean values is the same in both situations. In figure 1A, the SD values are low. Hence, the samples do not overlap and comparison using a hypothesis test yields a significant difference. In contrast, in figure 1B, the SD values are high and the samples thus overlap. In this circumstance, it is not possible to show a significant difference between the two samples using hypothesis tests.16
Figure 1.
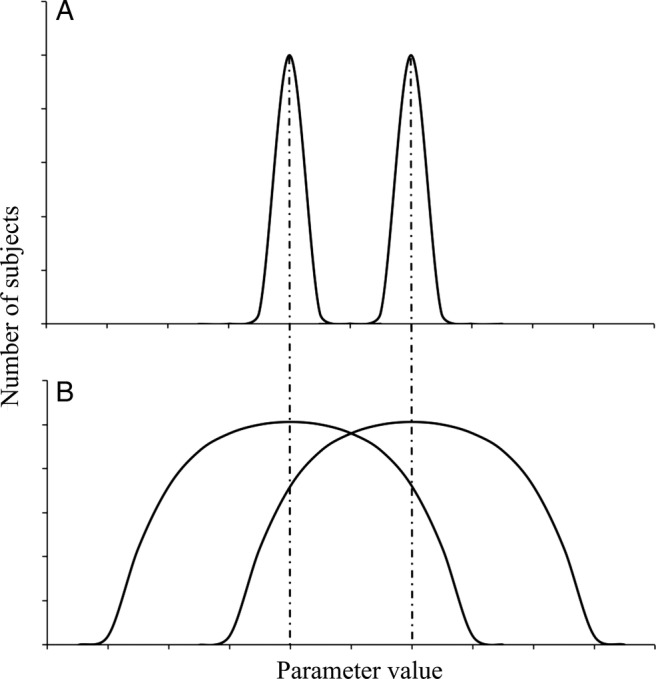
Two comparisons contrasting of two samples each by hypothesis tests, role of SD values. (A) Comparison of two samples with low SDs. (B) Comparison of two samples with wide SDs. In both cases (A and B), the difference between means is the same.
Hypothesis tests are used to demonstrate superiority of one treatment over another and are not valid to demonstrate equivalence. In equation 1, if n is small, the value of s is larger. Accordingly, it is impossible to show a significant difference between two samples, if the number of patients to be compared is small, because of high-SD values.15 16 This, of course, does not imply that the two samples being compared are equivalent.15 Therefore, clinical studies with small sample sizes are inadequate to demonstrate biosimilarity and can be misleading. To demonstrate that two treatments are equivalent, a clinical trial typically would require a larger sample size than if it were intended to demonstrate superiority of one over the other.
Equivalence analysis is less familiar to most clinicians than is hypothesis testing. However, the principles of equivalence analysis are not difficult to understand. The first consideration is that equivalence does not imply identity, but, rather, means that the results observed with a test product are within a pre-established range in relation to those obtained with the reference product.13 17 The limits of this equivalence range are arbitrary and are established on the basis of past experience. For a candidate biosimilar that is being developed to treat rheumatological diseases, the equivalence range typically is based on a meta-analysis of placebo-controlled clinical trials of the reference product. The mean difference in the outcome measure of interest between the reference product and placebo is calculated, and then divided by an arbitrary factor (usually of 2) to narrow the equivalence range.13 Once this range has been established, the clinical trial comparing the biosimilar to its reference product is conducted, the observed difference between treatments is assessed, and the 95% CIs of this difference are calculated. If these 95% CI are contained within the pre-established equivalence range, the two treatments are equivalent.13 17
How is equivalent efficacy determined in practice for a biosimilar compared to its reference (or innovator) product? As an example, let us consider the evaluation of an infliximab biosimilar in comparison to innovator infliximab (Remicade).13 18 First, a suitable patient population is chosen (ie, patients with rheumatoid arthritis taking methotrexate). Next, a suitable primary end point and treatment duration are chosen (ie, percentage of patients achieving the ACR20 response at week 30). Finally, an equivalence range is chosen. Since the difference in the ACR20 response between treatment with infliximab and placebo is about 30% in the several placebo-controlled clinical trials of infliximab treatment for rheumatoid arthritis, an equivalence range of half that value (15%) is considered to be reasonable. This means that the ACR20 response to treatment with the biosimilar can vary by ±15% with respect to that to treatment with the innovator. A logarithmic transformation of the data is then performed. With log-transformed data, the ±15% equivalence range is 0.85–1.18, so as to be symmetrical.
Now, let A and B represent mean values of an efficacy parameter for the candidate biosimilar and the innovator, respectively. The hypothesis to prove is shown in equation 2:
| 2 |
Once A and B have been determined for the two groups of treated patients studied, A/B is calculated and the corresponding 95% CIs are estimated. If the 95% CI for A/B lies entirely within the 0.85–1.18 range, the two treatments are considered to be equivalent. If this 95% CI exceeds the lower, higher, or both equivalence limits, the treatments are not considered to be. This is shown graphically in figure 2. White bars depict various 95% confidence limits for values of A/B that correspond to equivalence. The mean values can be low or high, and variability can be small or large. However, as long as the 95% CI lies within the pre-established range, equivalence of the two treatments is established. In each of these situations, the non-innovator is considered to be biosimilar to the innovator. Dark bars depict other 95% CIs for values of A/B that do not meet the definition of equivalence. Therefore, the candidate non-innovator biopharmaceutical cannot be considered to be a biosimilar. In each of these cases, the 95% CI goes beyond the lower, higher or both equivalence limits. The absolute magnitude of A/B does not matter when the 95% confidence limit exceeds either the lower or upper limit of the pre-established range. In that situation, equivalence cannot be established and the candidate non-innovator biopharmaceutical tested is considered to not be biosimilar to the reference product.17
Figure 2.

Graphical representation of equivalence analyses. (A and B) are the efficacy parameters of biosimilar and innovator, respectively. Bars correspond to the 95% CIs of A/B. Equivalence is declared when the entirety of the 95% CI is comprised within the equivalence interval (white bars). Equivalence cannot be declared when the 95% CI goes beyond the inferior, the superior or both equivalence limits (black bars).
The infliximab biosimilar CT-P13 showed efficacy equivalent to that of innovator infliximab (Remicade) in the PLANETRA study.18 In this study, 606 patients with rheumatoid arthritis, inadequately responsive to methotrexate, were randomised 1:1 to receive either CT-P13 or innovator infliximab. The primary efficacy outcome was the proportion of subjects achieving an ACR20 response at 30 weeks. At this time point, the response rates for the biosimilar and the innovator were 60.9% (184/302) and 58.9% (178/304), respectively, in the intention-to-treat population. The difference between treatments was 2% with a CI 95% of −6% to 10%. Since the entirety of this CI lies within the pre-established range of ±15% (0.85–1.18 after logarithmic transformation), the efficacy of CT-P13 was considered to be equivalent to that of innovator infliximab. A similar finding was observed in the per protocol analysis, where the response rates for the biosimilar and the innovator were 73.4% (182/248) and 69.7% (175/251), respectively. The difference between treatments in this analysis was 4%, with a 95% CI of −4% to 12%. Since this 95% CI also lies entirely within the pre-established range of ±15%, the two treatments again were considered to be equivalent.
Non-inferiority approach
In some circumstances, a non-inferiority approach can be suitable to evaluate biosimilarity.17 19 The guidelines on biosimilar evaluation issued by the WHO consider a non-inferiority design to be acceptable.20 Accordingly, several countries will accept such a non-inferiority approach. In Canada, regulatory authorities have acknowledged that an equivalence trial design is preferred. However, if clearly justified, a non-inferiority approach may be acceptable under certain conditions. Evidence must be provided that an eventual superiority has no clinical meaning and that there is no increase in adverse reactions with regard to the reference product.21 In comparative effectiveness clinical trials designed using a non-inferiority approach, the test product can be superior to the reference product but it cannot be inferior. If a biosimilar is to be compared to an innovator using a non-inferiority approach, the hypothesis to be proven is shown in equation 3:
| 3 |
In this situation, all of the 95% CI for log-transformed data must be above the inferiority limit. However, here there is no superiority limit. Examples of this are depicted in figure 3. White bars represent 95% CIs for values of A/B that correspond to non-inferiority. The results may be close to or far above the inferiority limit and variability can be small or large. However, as long as the inferiority limit is not exceeded, the biosimilar is considered to be non-inferior to the reference product. Alternatively, the dark bars represent values of A/B that do not meet the definition of non-inferiority, since the 95% CIs for these values of A/B go below the inferiority limit, regardless of whether or not variability is low or high.
Figure 3.

Graphical representation of non-inferiority analyses. (A and B) are the efficacy parameters of biosimilar and innovator, respectively. Bars correspond to the 95% CIs of A/B. Non-inferiority is declared when the entirety of the 95% CI lies above the inferiority limit (white bars). Non-inferiority cannot be declared when the 95% CI goes beyond the inferiority limit.
The non-inferiority approach has the advantage of requiring fewer patients to achieve statistical power.17 19 However, it is not adequate to assess biosimilarity of biopharmaecuticals used to treat rheumatological diseases, since it does not detect the situation in which a non-innovator is superior to the reference product and thus is bio-better and not biosimilar. A bio-better that produces a greater pharmacological response in some patients might have not only superior efficacy, but also increased toxicity.8 To date, no biosimilar has been approved using a non-inferiority approach to compare effectiveness to its reference product.
Other biosimilars
Presently, only one biosimilar has been approved by the EMA for use in rheumatology: the biosimilar infliximab CT-P13 (Remsima/Inflectra).2 This biosimilar monoclonal antibody has also been approved in Canada, Japan, Turkey, Colombia and South Korea,3 although with differences in the indications for which it has been authorised.4 CT-P13 has been submitted for review by the US Food and Drug Administration (FDA).3 There is worldwide interest regarding how the US FDA will consider this biosimilar. Will full extrapolation of indications be granted? Will additional clinical studies be requested? Such questions will soon be answered when the US FDA makes public its findings.
Other biosimilars to treat rheumatological diseases, including infliximab, adalimumab and etanercept, have been approved recently in India and South Korea.5–7 Other biosimilars are under development, many of which have been tested in clinical trials, and likely will reach the market over the coming years.2
The results of several trials of biosimilars in rheumatological diseases have been presented at major international rheumatology meetings. Bae et al22 compared an etanercept biosimilar, HD203, with innovator etanercept, Enbrel, in South Korean patients with rheumatoid arthritis inadequately responsive to methotrexate. A total of 294 patients were randomised 1:1 to receive either HD203 or innovator etanercept, each in combination with methotrexate. The primary end point was the proportion of patients achieving ACR20 response at week 24. Patients also were assessed at week 48. The difference in the ACR20 response rate between patients treated with HD203 and innovator etanercept was 2.12% with a 95% CI of −7.65% to 11.89% after 24 weeks, and was 4.37% with a CI of −5.57% to 14.31% after 48 weeks. Equivalence was concluded, although the published abstract did not disclose the pre-established equivalence range. On the basis of this trial, HD203 was approved in South Korea.7 Whether regulatory agencies in other countries will request additional information before approving this biosimilar etanercept remains to be seen.
Kay and coworkers compared BOW015, an infliximab biosimilar, with innovator infliximab (Remicade) in patients with rheumatoid arthritis inadequately responsive to methotrexate. A total of 189 patients were randomised 2:1 to receive BOW015 or innovator infliximab. The primary end point was the proportion of patients achieving an ACR20 response at week 16, with an equivalence margin of±23%. Data have been reported through 54 weeks.23 24 The differences in the ACR20, ACR50 and ACR70 response rates between patients treated with BOW015 and innovator infliximab were within this equivalence range, both by intention-to-treat and per protocol analysis. Furthermore, both biopharmaceuticals exhibited similar kinetics of these responses at several time points before the plateau phase of the time-response curve. The authors concluded therapeutic equivalence and BOW015 was recently approved as a biosimilar in India on the basis of this clinical trial.5 Whether other countries will grant approval to BOW015 as an infliximab biosimilar with the presently available clinical trial data remains to be seen.
As is evident, there is no standardised clinical assay for biosimilarity in rheumatological diseases at present. To date, studies of biosimilars to treat rheumatoid arthritis have used the ACR20 response as the primary end point. However, these clinical trials vary in their randomisation strategies, equivalence ranges and duration of treatment. Since additional biosimilars are being developed, the design of clinical trials to gain regulatory approval should be standardised.
Biomimics or intended copies
Biomimics, also known as intended copies, are non-innovator biologics that had received marketing approval before biosimilar regulations were put in place. They currently are available in some Asian and Latin American countries. Biomimics cannot be considered to be biosimilars, since they have not been subjected to review by an appropriate regulatory body according to a prespecified regulatory pathway for biosimilar approval.25 Limited and flawed clinical information is available regarding biomimics used to treat rheumatological diseases.
Yisaipu is an etanercept biomimic that is manufactured and marketed in China. It is also commercialised as Etanar in Colombia, as Etart in Mexico and as Etacept in India.2 8 12 25 26 Clinical experience in China has found Yisaipu to be effective.26 However, it is surprising that, despite this product having been used in China for over a decade, no data have been published about drug survival or on the incidence of tuberculosis and other adverse effects. Furthermore, no head-to-head studies have been conducted to comparing Yisaipu to innovator etanercept. Thus, this product cannot be considered to be an etanercept biosimilar. An abstract describing an open-label study of Etanar treatment in 110 patients with rheumatoid arthritis was presented at the 2010 ACR Annual Scientific meeting.27 The patients enrolled were receiving a variety of antirheumatic drug regimens concomitantly with Etanar. The number of patients studied was small, considering the variety of treatment regimens that were allowed. Etanar was not compared to innovator etanercept.
Infinitam is a non-innovator etanercept that is manufactured and marketed by the Mexican company, Probiomed.25 An abstract describing a study comparing Infinitam to innovator etanercept (Enbrel) was presented in the 2013 EULAR Annual Scientific meeting.28 The design of this study is confusing. Three groups of patients were studied. In the first group, 12 patients received Infinitam and methotrexate for 24 weeks. In the second group, 12 patients initially received Infinitam and methotrexate for 12 weeks, followed by innovator etanercept and methotrexate for the subsequent 12 weeks. Patients in these two groups were participating in a pharmacokinetic study. In the third group, 30 patients received Infinitam and methotrexate for 24 weeks. End points were DAS28 at weeks 12 and 24, but a primary end point was not specified. The authors stated that DAS28 improved and that drug safety was similar in all treatment groups. However, it appears that only patients enrolled in the first and third groups received the same treatment regimen. The only distinction between these groups was that patients in the first group were participating in a pharmacokinetic study, whereas those in the third group were not. The authors concluded that “none [sic] significant difference was observed in the pharmacokinetic groups (p=0.355)”. As mentioned earlier, failure to detect a significant difference is evidence of neither equivalence nor of biosimilarity. This is particularly true when the sample size is evidently small (12 patients per group), yielding high-SD values. Thus, based on this study, Infinitam cannot be considered to be an etanercept biosimilar. Whereas the primary reason for a biosimilar to be used instead of the innovator is its lower price, the cost of this biomimic in some acquisition contracts by the Social Security in Mexico has been the same as that of innovator etanercept.25
As can be appreciated, there is an enormous difference in quality between the studies described in the abstracts about the biosimilars HD203 and BOW015, and those about the biomimics Etanar and Infinitam. The data on HD203 and BOW015 likely will be published in peer-reviewed journals in the near future, allowing readers to assess the advantages as well as the limitations of these biosimilars. On the other hand, no peer-reviewed publication documents the efficacy or safety of Yisaipu/Etanar/Etart or of Infinitam, despite these products having been available and used to treat patients with rheumatological diseases in several countries for several years.
Kikuzubam is a rituximab biomimic that was manufactured and marketed in Mexico by Probiomed.2 8 25 In 2012, the Mexican programme of Pharmacovigilance issued a communication to health professionals warning them of anaphylactic reactions that occurred in several patients who were switched from innovator rituximab (Mabthera) to the biomimic, or vice versa.29 This was surprising, since innovator rituximab had exhibited a very favourable peri-infusion safety profile among Mexican patients.30 Owing to these anaphylactic reactions and the lack of clinical data documenting the efficacy and safety of Kikuzubam, approval to market Kikuzubam in Mexico was withdrawn by the regulatory authority on 28 March 2014.31
Reditux is a rituximab biomimic manufactured in India and marketed in India and in several Latin American countries.2 8 To the best of our knowledge, no clinical trial has been performed to demonstrate equivalent efficacy and safety of Reditux with innovator rituximab (Mabthera) in patients with a rheumatological disease. However, analytical studies have demonstrated significant differences in physicochemical properties between Reditux and innovator rituximab.32 Thus, Reditux cannot be considered to be a rituximab biosimilar.
Concluding remarks
True biosimilars should not be confused with biomimics that have not undergone rigorous clinical testing or regulatory review according to a pathway for approval of biosimilars. There are only two options for biomimics: either document their efficacy and safety in well-designed comparative effectiveness clinical trials, and subject them to regulatory review so that they can become true biosimilars, or remove them from the market altogether.
The perception of biosimilars in rheumatology has evolved over the past several years. Distrust about the use of biosimilars in clinical practice has waned as regulatory pathways have been implemented and applied to the approval of a biosimilar infliximab. Although analytical and non-clinical evidence plays a considerable role, clinical trials that demonstrate comparable efficacy and safety of the biosimilar with its reference product are of paramount importance to establish biosimilarity. Familiarity of clinicians with issues regarding study design and the methods of analysing data obtained in comparative effectiveness clinical trials undoubtedly will improve decision-making surrounding the prescription of biosimilars to patients with rheumatological diseases.
Acknowledgments
The authors would like to thank Dr Ricardo Garcia (CLAPBio) for his useful comments and suggestions.
Footnotes
Contributors: GC-H, RG-R and MAS conceived and drafted, this manuscript. GC-H, RG-R, JK and MAS revised this review critically for important intellectual content and approved the version submitted.
Funding: This work was supported by CINVESTAV and CLAPBio.
Competing interests: GC-H has received consultancy fees from Amgen, Abbvie, Astra-Zeneca, Bayer, Boehringer-Ingelheim, Eli-Lilly, Janssen-Cilag, Laboratorios Liomont, Laboratorios Sophia, Medix, Merck-Serono, Merck, Sharp and Dohme, Novartis, Pfizer, Roche, Sanofi and UCB. RGR has received consultancy fees from Amgen and Sanofi. JK has received research support (paid to the University of Massachusetts Medical School) from AbbVie Inc; Ardea Biosciences, Inc; Eli Lilly and Company; Pfizer Inc; and Roche Laboratories, Inc; and has received consultancy fees from Alexion Pharmaceuticals; Amgen, Inc; AbbVie Inc; AstraZeneca; Boehringer Ingelheim GmbH; Bristol-Myers Squibb Company; Crescendo Bioscience, Inc; Eli Lilly and Company; Epirus Biopharmaceuticals, Inc; Genentech Inc; Hospira, Inc; Janssen Biotech, Inc; Merck Sharp and Dohme Corp; Nippon Kayaku Co, Ltd; Novartis Pharmaceuticals Corporation; PanGenetics, BV; Pfizer Inc; Samsung Bioepis; Roche Laboratories, Inc; and UCB, Inc. MAS has received consultancy fees from Abbvie, Bioepis, Boehringer Ingelheim, Epirus, Janssen, Orygen, Pfizer and Samsung.
Ethics approval: This is a review article. No ethical approval was needed.
Provenance and peer review: Commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Scheinberg MA, Kay J. The advent of biosimilar therapies in rheumatology—“O brave new world”. Nat Rev Rheumatol 2012;8:430–6. 10.1038/nrrheum.2012.84 [DOI] [PubMed] [Google Scholar]
- 2.Castañeda-Hernández G, Szekanecz Z, Mysler E et al. . Biopharmaceuticals for rheumatic diseases in Latin America, Europe, Russia, and India: innovators, biosimilars, and intended copies. Joint Bone Spine 2014;81:471–7. 10.1016/j.jbspin.2014.03.019 [DOI] [PubMed] [Google Scholar]
- 3.Celltrion. About us. What's new. http://www.celltrion.com/en/company/notice_view.asp?idx=456&code=ennews&intNowPage=1&menu_num=&align_year=all (accessed 18 Apr 2015).
- 4.Feagan BG, Choquette D, Ghosh S et al. . The challenge of indication extrapolation for infliximab biosimilars. Biologicals 2014;42:177–83. 10.1016/j.biologicals.2014.05.005 [DOI] [PubMed] [Google Scholar]
- 5.GaBi Online—Generics and Biosimilars Initiative. Infliximab “similar biologic” receives Indian approval. http://www.gabionline.net/layout/set/print/Biosimilars/News/Infliximab-similar-biologic-receives-Indian-approval (accessed 18 Apr 2015).
- 6.GaBi Online—Generics and Biosimilars Initiative. Adalimumab similar biologic launched in India. http://gabionline.net/Biosimilars/News/Adalimumab-similar-biologic-launched-in-India (accessed 18 Apr 2015).
- 7.Robinson R. HD203 biosimilar is clinically equivalent to etanercept. http://www.hcplive.com/conferences/acr-2014/HD203-Biosimilar-is-Clinically-Equivalent-to-Etanercept (accessed 18 Apr 2015).
- 8.Dörner T, Strand V, Castañeda-Hernández G et al. . The role of biosimilars in the treatment of rheumatic diseases. Ann Rheum Dis 2013;72:322–8. 10.1136/annrheumdis-2012-202715 [DOI] [PubMed] [Google Scholar]
- 9.Yi S, Kin SE, Park MK et al. . Comparative pharmacokinetics of HD203, a biosimilar of etanercept, with marketed etanercept (Enbrel®): a double-blind, single-dose, cross-over study in healthy volunteers. BioDrugs 2012;26:177–84. 10.2165/11631860-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 10.Kaur P, Chow V, Zhang N et al. . A randomized, single-blind, single-dose, three-arm, parallel group study in healthy subjects to demonstrate pharmacokinetic equivalence of ABP 501 and adalimumab: results of comparison with adalimumab (EU). Ann Rheum Dis 2014;73(Suppl 2):479 [Abstract] 10.1136/annrheumdis-2014-eular.2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Araújo F, Cordeiro I, Teixeira F et al. . Pharmacology of biosimilar candidate drugs in Rheumatology: a literature review. Acta Rheumatol Port 2014;39:19–26. [PubMed] [Google Scholar]
- 12.Scheinberg MA, Azevedo VF. Biosimilars in rheumatology: perspectives and concerns. Rheumatology (Oxford) 2014;53:389–90. 10.1093/rheumatology/ket210 [DOI] [PubMed] [Google Scholar]
- 13.Kay J, Smolen JS. Biosimilars to treat inflammatory arthritis: the challenge of proving identity. Ann Rheum Dis 2013;72:1589–93. 10.1136/annrheumdis-2012-203198 [DOI] [PubMed] [Google Scholar]
- 14.Cortés J, Curigliano G, Diéras V. Expert perspectives on biosimilar monoclonal antibodies in breast cancer. Breast Cancer Res Treat 2014;144:233–9. 10.1007/s10549-014-2879-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pidgen AW. Statistical aspects of bioequivalence—a review. Xenobiotica 1992;22:881–93. 10.3109/00498259209053147 [DOI] [PubMed] [Google Scholar]
- 16.Patel BV. Bioequivalence: an overview of statistical concepts. Indian J Pharmacol 2004;36:209–16. [Google Scholar]
- 17.Njue C. Statistical considerations for confirmatory clinical trials for similar biotherapeutic products. Biologicals 2011;39:266–9. 10.1016/j.biologicals.2011.06.006 [DOI] [PubMed] [Google Scholar]
- 18.Yoo DH, Hrycaj P, Miranda P et al. . A randomized, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis 2013;72:1613–20. 10.1136/annrheumdis-2012-203090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fletcher M. Biosimilars clinical development program: confirmatory clinical trials: a virtual/simulated case study comparing equivalence and non-inferiority approaches. Biologicals 2011;39:270–7. 10.1016/j.biologicals.2011.06.004 [DOI] [PubMed] [Google Scholar]
- 20.World Health Organization Expert Committee on Biological Standardization. Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs). Geneva, 19–23 October 2009. [http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf (accessed 18 Apr 2015).
- 21.Canadian Agency for Drugs and Technologies in Health. Subsequent Entry Biologics—Emerging Trends in Regulatory and Health Technology Assessment Frameworks. https://www.cadth.ca/subsequent-entry-biologics-emerging-trends-regulatory-and-health-technology-assessment-frameworks (accessed 18 Apr 2015).
- 22.Bae SC, Kim JS, Choe JY et al. . A randomized, double-blind, phase 3, equivalence trial comparing the etanercept biosimilar, HD203, with etanercept (Enbrel®), in combination with methotrexate (MTX) in patients with rheumatoid arthritis (RA) [Abstract]. Arthitis Rheumatol 2014;66:2825. [Google Scholar]
- 23.Kay J, Chopra A, Chandrashekara S et al. . A phase 3, randomized, double-blind, active comparator study of the efficacy and safety of BOW015, a biosimilar infliximab, in patients with active rheumatoid arthritis on stable methotrexate doses. Ann Rheum Dis 2014;73(Suppl 2):64. [Abstract]. [Google Scholar]
- 24.Kay J, Wyand M, Chandrashekara S et al. . BOW015, a biosimilar infliximab, in patients with active rheumatoid arthiritis on stable methotrexate doses: 54-week results of a randomized, double-blind, active comparator study [Abstract]. Arthitis Rheumatol 2014;66:3538. [Google Scholar]
- 25.Scheinberg M, Castañeda-Hernández G. Antitumor necrosis factor patent expiration and the risks of biocopies in clinical practice. Arthritis Res Ther 2014;16:501 10.1186/s13075-014-0501-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kay J. Biosimilars: a regulatory perspective from America. Arthritis Res Ther 2011;13:112 10.1186/ar3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rondón F, Bautista A, Salazar JC et al. . Etanar therapy in real-life patients with rheumatoid arthritis. Arthritis Rheum 2010;62(Suppl 10):1811. [Google Scholar]
- 28.Moctezuma JF, Martínez A, Enkerlin H et al. . Comparative, randomized, simple blind to evaluate efficacy and safety of Infinitam® (etanercept), associated with methotrexate compared with Enbrel® (etanercept) associated with methotrexate in patients with moderate and severe rheumatoid arthritis. Ann Rheum Dis 2013;72(Suppl 3):A234. [Abstract]. 10.1136/annrheumdis-2013-eular.736 [DOI] [Google Scholar]
- 29.Comisión Federal para la Protección contra Riesgos Sanitarios. Comunicado a los Profesionales de la Salud. Reacciones anafilácticas por el uso de Rituximab. 4 de abril de 2012. http://www.cofepris.gob.mx/AZ/Paginas/Farmacovigilancia/Comunicados.aspx (accessed 18 Apr 2015).
- 30.Arredondo-Garza T, Majluf-Cruz A, Vela-Ojeda J, Peri-infusional adverse reactions to rituximab in patients with non-Hodgkin's lymphoma. Arch Med Res 2013;44:549–54. 10.1016/j.arcmed.2013.09.011 [DOI] [PubMed] [Google Scholar]
- 31.Comisión Federal para la Protección contra Riesgos Sanitarios. Alerta Sanitaria: La COFEPRIS revoca registro de producto “Kikuzubam”. 28 de marzo de 2014. http://www.google.com.mx/url?sa=t&rct=j&q=&esrc=s&source=web&cd=3&ved=0CCwQFjAC&url=http%3A%2F%2Fwww.cofepris.gob.mx%2FAS%2FDocuments%2FCOMISI%25C3%2593N%2520DE%2520OPERACI%25C3%2593N%2520SANITARIA_Documentos%2520para%2520publicar%2520en%2520la%2520secci%25C3%25B3n%2520de%2520MEDICAMENTOS%2FAlertas%2FKIKUZUBAM_28032014_.pdf&ei=D3LNVJT3K5O3yAS_0ILgCg&usg=AFQjCNE3DGEjuIq7l8heQHqbq7PXgkITiA&sig2=_QtwfdJhjkihHbjvHDPZEg&bvm=bv.85076809,d.aWw (accessed 18 Apr 2015).
- 32.Espinoza de la Garza CE, Perdomo-Abúndez FC, Padilla-Calderón J et al. . Analysis of recombinant monoclonal antibodies by capillary zone electrophoresis. Electrophoresis 2013;34:1133–40. 10.1002/elps.201200575 [DOI] [PubMed] [Google Scholar]