

Mutant Ras is one of the most prevalent oncogenes in human cancers. The ability of oncogenic Ras to promote tumor growth in a cell intrinsic manner has been appreciated. In addition to hyperplasia, activated Ras controls cancer cell differentiation status marked by metaplasia, epithelial-mesenchymal transition, and acquisition of stem-cell traits. Over the last decade, the ability of mutant Ras to promote tumor development and progression in a non-cell autonomous manner has been unraveled. Through the up-regulation and secretion of various cytokines and chemokines, mutant Ras has been shown to directly contribute to malignancy by promoting basement membrane degradation and dysplasia, neovascularization, and suppression of anti-tumor immunity. However, the exact mechanism through which mutant Ras elicits the pro-inflammatory secretory response remains unknown. In 2 recent studies, we report squamous cell carcinoma antigens (SCCA) SerpinB3/SerpinB4 as novel pro-tumorigenic mediators downstream of mutant Ras via the activation of NF-κB and pro-inflammatory cytokine production.1,2
SCCAs are members of the serpin family of endogenous serine/cysteine proteinase inhibitors. The first variant of SCCAs, SCCA1 (SerpinB3), was originally identified in squamous cell carcinoma (SCC) of the uterine cervix.3 Unlike many other serpins such as plasminogen activator inhibitors (PAI) that function extracellularly, SCCAs belongs to the “Clade B” subfamily of the intracellular serpins that are believed to mostly inhibit cathepsins.4 Elevated SCCA expression has been found in many human cancers and is associated with poorly differentiated, inflammatory, and more aggressive human breast cancer and hepatocellular carcinoma.5-7 Its tumor-promoting function has been mostly attributed to its anti-apoptotic activity against various stress/damaging conditions. However, while the anti-cell death function certainly supports SCCA1's pro-tumorigenic role, it does not seem to be sufficient in explaining SCCA1's association with the poorly-differentiated and more aggressive cancers. Attempting to address this, in the Sheshadri et al. work,2 we report that ectopically expressed SCCA1 drives epithelial-mesenchymal transition (EMT) as well as oncogenic transformation in a number of epithelial cells including the non-transformed mammary epithelial cell line MCF10A. This is through a cell-autonomous activation of NF-κB and the production of pro-inflammatory cytokines such as IL-6. The ability of SCCA to promote NF-κB activation and cytokine production appears to be dependent on its capacity to disrupt protein degradation pathways and trigger a non-lethal unfolded protein response (UPR). The UPR effectors, ATF6α and PERK, are activated and are essential in maintaining SCCA-induced cytokine production and transformation. Consistent with these observations made in cultured cells, exaggerated stress response associated with an inflammatory stromal reaction is also observed in vivo when a mammary specific transgenic mouse model of SCCA1 is crossed to the MMTV-Neu mice.
In a parallel study to address the molecular regulation of SCCA, we find that SCCA1 and SCCA2 are transcriptional targets of mutant Ras, and that they play an essential role in Ras-induced pro-inflammatory conditions and oncogenesis.1 Introduction of mutant Ras results in a robust transcriptional increase in SCCA1 and SCCA2 in multiple cell lines. This Ras-driven upregulation of SCCA is dependent on the MAPK/ERK arm and the ETS family transcription factor PEA3. The positive correlation between Ras mutations and increased SCCA expression is observed in human colorectal cancer samples. In human pancreatic cancer where mutant K-Ras is the predominant driver mutation, SCCA is up-regulated in the earliest pancreatic intraepithelial neoplasia (PanIN) lesions with its positivity increasing through PanIN progression and pancreatic ductal adenocarcinoma (PDA) development. As mutant Ras is known to promote NF-κB activity and pro-inflammatory cytokine production, SCCA silencing results in a robust decrease in the production and secretion of numerous cytokines accompanied by ablated xenograft tumor growth in Ras-activated pancreatic cancer cell lines. Consistent with the observations in mammary epithelial cells, the ability of SCCA to promote NF-κB activation and cytokine production appears to be dependent on SCCA's ability to induce UPR. Along these lines, independent silencing of the ATF6 and XBP1 arms of the UPR diminishes Ras-induced cytokine production, whereas ectopic expression of the active spliced XBP1 restores cytokine production following SCCA silencing.
Our findings implicate SCCA in pro-inflammatory cytokine production in Ras-driven tumors through a UPR-dependent manner (Fig. 1). Thus by regulating the inflammatory circuit, SCCA may control both autocrine pro-tumorigenic signals as well as paracrine remodeling of the tumor landscape. While SCCA's ability to disrupt protein degradation pathways has been well demonstrated, the precise mechanism by which SCCA induces UPR remains unknown. Identification of SCCA interacting partners and subcellular localization will help in addressing this question. Nonetheless, our studies indicate that SCCA's protease inhibitory function is crucial to its function in inducing UPR and thus cytokine production and oncogenesis. Inhibition of SCCA's protease inhibitory ability may be a vital therapeutic option in cancers driven by mutant Ras.
Figure 1.
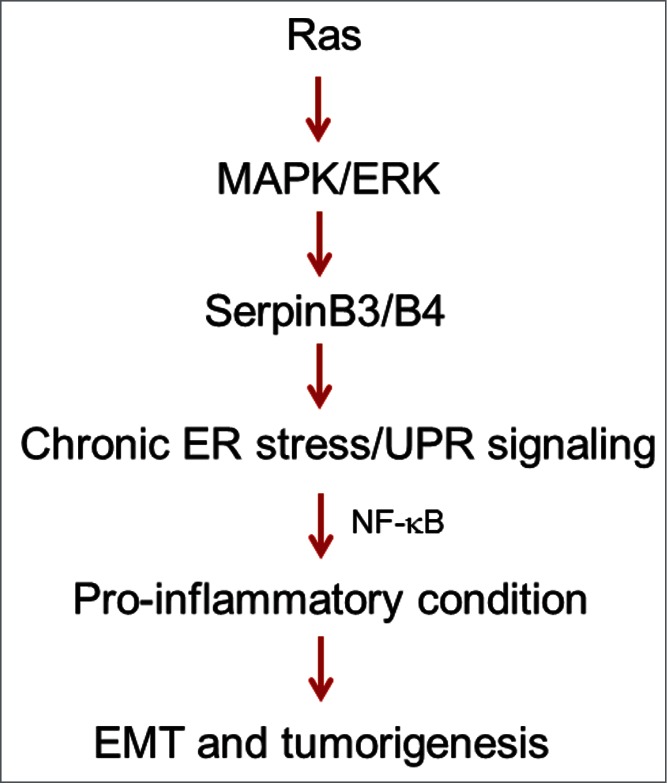
Diagram of SCCA1/2 (SerpinB3/B4) as critical mediators in Ras-driven cancers. Mutant Ras induces SCCA SerpinB3/B4 expression via the MAPK/ERK/PEA3 pathway. SCCA induces UPR and subsequent activation of NF-κB and the production of inflammatory cytokines such as IL-6. This leads to cytokine autocrine signaling as well as inflammatory conditions that promotes EMT and oncogenesis.
References
- 1. Catanzaro JM, et al. Nat Commun 2014; 5:3729; PMID:24759783; http://dx.doi.org/ 10.1038/ncomms4729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sheshadri N, et al. Cancer Res 2014; canres.0798.2014; PMID:2521332225213322 [Google Scholar]
- 3. Kato H, et al. Cancer 1977; 40:1621-28; PMID:332328; http://dx.doi.org/ [DOI] [PubMed] [Google Scholar]
- 4. Schick C, et al. Biochemistry 1998; 37:5258-66; PMID:9548757; http://dx.doi.org/ 10.1021/bi972521d [DOI] [PubMed] [Google Scholar]
- 5. Catanzaro JM, et al. PLoS One 2011; 6:e19096; PMID:21526154; http://dx.doi.org/ 10.1371/journal.pone.0019096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Collie-Duguid ES, et al. Breast Cancer Res Treat 2011; PMID:21695460 [DOI] [PubMed] [Google Scholar]
- 7. Turato C, et al. Eur J Cancer 2012; 48:1219-26; PMID:21737255; http://dx.doi.org/ 10.1016/j.ejca.2011.06.004 [DOI] [PubMed] [Google Scholar]