

Orchestrating the multiple enzymatic activities required at the eukaryotic replication fork would appear to be a complex task. The proliferating cell nuclear antigen (PCNA) has long been recognized as a key contributor to this process, binding a large diversity of partner proteins and recruiting them to their sites of action.1 PCNA partners recruited to replication forks include DNA polymerases, Okazaki fragment processing enzymes, DNA methyltransferases, cell cycle regulators, and chromatin assembly and modifying enzymes, each required at different stages of chromosomal replication. The importance of PCNA for this most crucial of cellular functions is underlined by the fact that, until recently, no disease causing mutation in PCNA had been reported in humans, suggesting that most sequence alterations that affect PCNA function would be incompatible with life.
We identified a neurodegenerative condition among the Ohio Amish community, with features overlapping with known disorders of DNA repair.2 Affected individuals exhibit sunlight sensitivity, photophobia and short stature somewhat reminiscent of xeroderma pigmentosum and Cockayne syndrome, and ocular and cutaneous telangiectasia indistinguishable from those seen in ataxia telangiectasia patients. Prelingual onset of sensorineural hearing loss, a feature not commonly associated with other DNA repair disorders, was universally seen. All affected individuals were found to be homozygous for a c.683G>T Amish founder mutation in PCNA, which results in the substitution of a stringently conserved serine at position 228 of the protein, to isoleucine (p.Ser228Ile). PCNA is an essential gene, yet individuals who are homozygous for this amino acid substitution survive, therefore the p.Ser228Ile substitution must not completely destroy the function of the PCNA protein. Consistent with this, our studies did not detect any changes in the essential process of DNA replication in cells from affected individuals. However, we were able to identify altered parameters of DNA repair, specifically within the nucleotide excision repair (NER) pathways, that deal with damage to DNA generated by sunlight exposure.3 PCNA is important for repair synthesis in NER,4 hence this finding provides a clear molecular link between the disease causing genetic alteration and the cellular phenotypes observed.
Further clarification of the consequences of the mutation was provided by our studies of the binding capabilities of the mutated PCNA protein. Many of the proteins recruited by PCNA to replication forks have additional roles in repair, including the DNA processing enzymes Ligase 1, and Flap endonuclease 1 (Fen1). PCNA also specifically associates with proteins with direct repair roles, such as the NER protein XPG. Our studies revealed that the interactions between PCNA and Fen1, Ligase 1 and XPG were significantly perturbed by the p.Ser228Ile substitution. It seems likely that this perturbation contributes to the defects in DNA repair pathways identified and to the clinical features seen in affected individuals.
While we were able to detect altered interaction between PCNA and 3 of its known binding partners, these are unlikely to represent the only outcomes of the sequence alteration. A systematic approach will be needed to assess the effect of the mutation on the binding to each known partner to ascertain whether other altered interactions may contribute to the clinical manifestations of the disease. While the interactions between PCNA and XPG, Ligase1 and Fen1 are mediated by PIP (PCNA interacting protein) boxes,5 our data suggest that not all PIP box containing proteins show altered affinity for PCNA p.Ser228Ile. For example the DNA methyltransferase DNMT1 apparently binds both wildtype and Ser228Ile PCNAs equivalently. Further work is needed to understand the structural changes triggered by the Ser228Ile amino acid alteration that result in such differential binding of some PCNA partners, but not others.
The separation of function outcome caused by this mutation is, as first sight, remarkable. PCNA's main role is during chromosomal replication, and yet the Amish individuals affected with this syndrome reach adulthood, and lymphoblastoid and primary fibroblast cells proliferate normally in culture with no apparent checkpoint activation or genomic instability. Thus the defective interactions observed, which are with key components of the DNA replication fork, appear not sufficient to significantly perturb replication while they do significantly impair DNA repair. This presents a conundrum as the part of the NER process that is presumed to be PCNA-dependent, DNA synthesis, is enzymatically the same in both situations. How can we reconcile this? We postulate that this may be explained by the nuclear organization of the 2 processes (replication and repair). Nucleotide excision repair is performed independently at every damage site, and these are distributed randomly across the genome. In contrast, DNA replication processes occur within ‘replication factories’ containing high local concentrations of DNA replication components, including PCNA, Fen1 and Ligase1.6 A single factory can contain multiple replication forks, and at each fork there are likely more than 2 PCNA trimers, as PCNA is retained for some time on newly synthesized DNA.6 Such a high local concentration of PCNA at replication factories may serve to allow PCNA's efficient interaction with partner proteins even if the affinity of such interactions is reduced, as with the p.Ser228Ile alteration (Fig. 1). In contrast, during NER there is no process that results in local concentration of PCNA, and as such the very same interactions may become sensitive to Ser228Ile mediated changes in interaction affinity.
Figure 1.
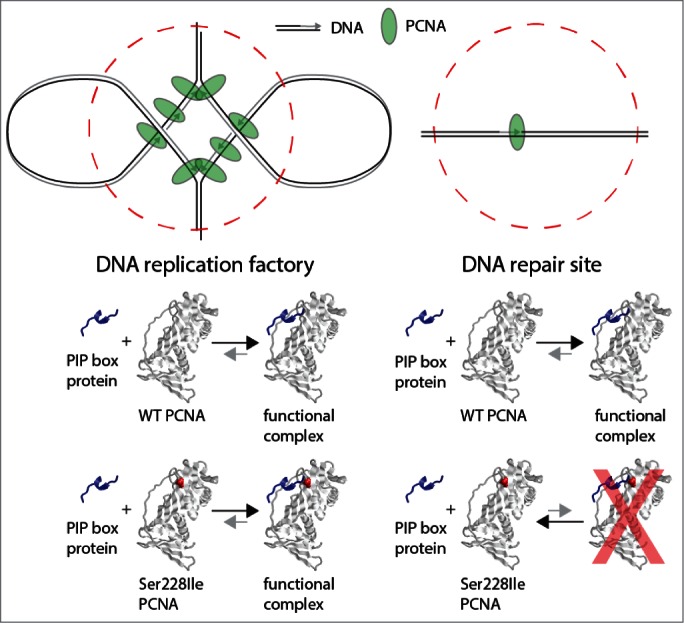
How can PCNA mutation affect DNA repair but not replication? Top: Schematic of DNA replication and repair sites in vivo. Replication factories processing Okazaki fragments generate high local concentrations of PCNA, not seen at repair sites. Bottom: The Ser228Ile alteration in PCNA destabilises its interaction with PIP-box containing partner proteins, increasing the KD for crucial binding reactions. This will have a greater biological effect when PCNA concentrations are low, i.e., at repair sites. Crystal structure of PCNA and PIP box rendered in Rasmol from 1AXC (www.rcsb.org).
This raises the intriguing possibility that the organization of replication into these factory structures may contribute to the stability of the replicative process, as the locally induced high concentrations of factors induced by such a physical organization make the system inherently robust against any temporary stresses. This could be predicted to be a useful property for a process in which accuracy is ultimately required to ensure the stability of the genome.
References
- 1. Mailand N, et al. . Nat Rev Mol Cell Biol 2013; 14(5):269-82; PMID:23594953; http://dx.doi.org/ 10.1038/nrm3562 [DOI] [PubMed] [Google Scholar]
- 2. Baple EL, et al. . J Clin Invest 2014; 124(7):3137-46; PMID:24911150; http://dx.doi.org/ 10.1172/JCI74593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. DiGiovanna JJ, et al. . J Invest Dermatol 2012; 132(3 Pt 2):785-96; PMID:22217736; http://dx.doi.org/ 10.1038/jid.2011.426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lehmann AR. DNA Repair (Amst) 2011; 10(7):730-3; PMID:21601536; http://dx.doi.org/ 10.1016/j.dnarep.2011.04.023 [DOI] [PubMed] [Google Scholar]
- 5. Warbrick E. Bioessays 1998; 20(3):195-9; PMID:9631646; http://dx.doi.org/ 10.1002/(SICI)1521-1878(199803)20:3%3c195::AID-BIES2%3e3.0.CO;2-R [DOI] [PubMed] [Google Scholar]
- 6. Sporbert A, et al. . Nucleic Acids Res 2005; 33(11):3521-8; PMID:15972794; http://dx.doi.org/ 10.1093/nar/gki665 [DOI] [PMC free article] [PubMed] [Google Scholar]