

Sir,
Single gene disorders are rare by themselves but collectively these are an important cause of morbidity and mortality. The identification of genes for single gene disorders has value, not only in prenatal diagnosis and genetic counselling of affected families, but also in basic research towards understanding gene functions and mechanisms of disease. The classical methods of gene identification, like chromosomal breakpoint mapping, linkage analysis and homozygosity mapping, are laborious and require multiple families with multiple number of affected individuals, thus are difficult to be used for single gene disorders occurring sporadically or exhibiting phenotypic heterogeneity1. On the other hand, the availability of targeted capture of exonic regions (exome), combined with massively parallel (next generation) sequencing technologies have made it possible to quickly and cost effectively identify genes and alleles responsible for a particular disease using just a few affected individuals2. In the present study we report a novel mutation in GJC2 gene that was identified by exome sequencing in two patients from a family with presumed single gene disorder causing slowly progressive ataxia and nystagmus with normal cognition.
The proband (patient A) was a five year old girl, who presented to the outpatient department of Medical Genetics at the Sanjay Gandhi Postgraduate Institute of Medical Sciences (SGPGIMS), Lucknow, India, in August 2011, with complaints of head bobbing noticed since five months of age. The child was born out of a consanguineous marriage. As she started to sit at 7-8 months she was unsteady. Subsequently, parents noticed tightness of lower limbs, broad based walk and inability to balance when she started to walk at 1.5 years of age. There was history of frequent falls while walking. She was not able to walk unaided at the time of examination. There was a history of straining of eyes and bringing objects close to eyes for seeing clearly. Head circumference was 48.5 cm corresponding to 30th centile for her age and gender. Spasticity was present in both lower limbs more than the upper limbs. Knee and ankle jerks were brisk. Cognition was normal but the speech was slow and unclear. There was no thinning of limbs or involuntary movements. Nystagmus, truncal ataxia and incordination of movements were present. Fundus examination showed bilateral optic atrophy. Magnetic resonance imaging (MRI) scan of brain revealed white matter hyperintensities involving central as well as peripheral subcortical white matter. No vacuolization was seen. Genu of corpus callosum was also involved. Internal capsule, pons and brachium pontis showed similar changes (Figure A). Metabolic analysis including gas chromatography mass spectrometry for organic acids, amino acid and acylcarnitine profile by tandem mass spectrometry was normal. Arylsulphatase A levels were normal. She had a similarly affected sister (patient B) who was two years younger than her. Her motor milestones were delayed and she could not sit without support. Hypertonia, nystagmus, truncal ataxia and incoordination of movements were present. The siblings had two other normal healthy brothers.
Figure A.
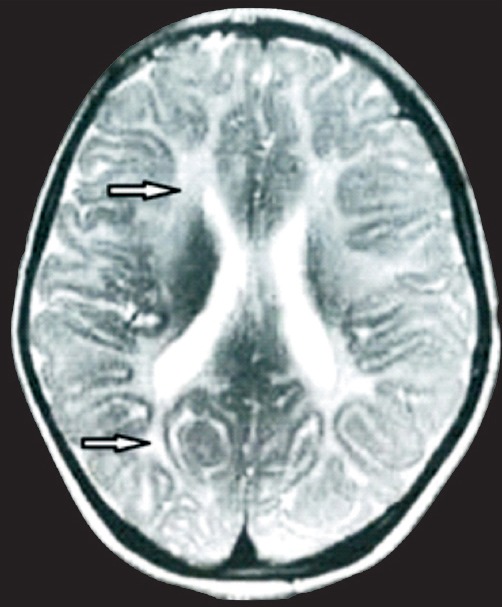
MRI image of patient A showing extensive leukodystrophy in frontal and occipital white matter (arrow).
Peripheral blood (5 ml) was collected in EDTA from the probands, their parents, normal brothers and a control sample from an unrelated healthy individual was used for genomic DNA isolation. Informed written consent was obtained from all the individuals participating in the study and the study was approved by the Institutional Ethics Committee of SGPGIMS, Lucknow. Microarray and exome sequencing followed by molecular analysis was done over a period of four months (December 2012 - March 2013). Single nucleotide polymorphism (SNP)-based microarray analysis of the patients A and B was performed using CytoScan 2.7M array (Affymetrix, USA). The Affymetrix CytoScan 2.7M array contains more than 2.3 million markers for copy number analysis and approximately 400,000 SNPs and data analysed using the software program Chromosome Analysis Suite version CytoB-N1.2.2.271 (Affymetrix, USA). Homozygosity mapping was done using gPLINK version 2.0503. Exome capture and sequencing was done on the DNA sample by outsourcing. Exome capture was performed on 3 µµg of genomic DNA sample from patient A using TargetSeq Exome Capture kit (Life Technologies, USA) and sequencing was done on SOLiD 5500xL platform following the manufacturer's instructions (Life Technologies, USA). Analysis of raw data obtained from exome sequencing was done at Centre for DNA Fingerprinting and Diagnostics, Hyderabad. The reads obtained were analysed using LifeScope version 2.5 software (Life Technologies, USA) by mapping against Human Genome Build 19, followed by detection of single nucleotide variants (SNVs) and small Indels. The variants identified were compared with those in NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi). Variant annotation was done using SeattleSeq version 8.074 for location and predicted function. Oligonucleotide primers were designed using Primer3 version 4.0.0 for amplification of the region of interest in GJC2 gene for capillary sequencing to confirm the findings of exome sequencing. Primer sequences were - Forward: 5’-TTTAAGGCGGTAAGCTCCAC-3’ and Reverse:5’TGCTCGTCCGAGTAGATGG-3’. Sanger sequencing were performed using ABI 3130 Genetic analyzer (Life Technologies, USA) following the manufacturer's protocol in patients, unaffected brothers, parents and a control sample. Sequencing data were analysed using Sequencing Analysis Software version 5.2 (Life Technologies, USA).
Array comparative genomic hybridization in the patients did not reveal any significant genomic deletion or duplication. However, multiple common LOH (loss of heterozygosity) regions were identified in the two patients, which were matched for the homozygous variants (SNPs and Indels) observed in the exome sequencing analysis. Exome sequencing of patient A, generated a total of 172348481 reads of which 116657472 reads were mapped to reference genome. Of these reads, 61.6 per cent reads were on target. Among the reads on target, coverage of minimum 20X was achieved for 89 per cent reads. Variant detection revealed a total of 42439 SNP variants and 2037 Indels. Of these, 1721 SNP variants and 66 Indels were absent in dbSNP. Among these candidate variants 643 SNP variants and 25 Indels were homozygous (Table). After stringent filtering (for SNPs, missense variations with PolyPhen score ≥ 0.5 and Grantham score ≥ 50 and for Indels, score PhastCons ≥ 0.5, were taken as a significant variant), none of the SNP variants were observed to fall in the overlapping homozygous regions detected by homozygosity mapping. At the same time five potential Indel variants were observed to be within the LOH regions in the affected girl, among which only one variant; a single base pair (G) deletion in GJC2 gene, was seen to be matching with the phenotype. After correlating the phenotype data with this mutation, it was identified as a probable candidate mutation for the condition in patient. This single base deletion identified in patient A by exome sequencing was confirmed by Sanger sequencing in both the patients (Figure B). Both parents were found to be heterozygous for the deletion (Figure B). The unaffected brothers were found to be heterozygous for the deletion. Whole exome sequencing analysis revealed a novel single bp (G) deletion in GJC2 gene which encodes a gap junction protein Cx47, implicated in intercellular communication. Earlier reports show that mutations in this gene lead to Pelizaeus-Merzbacher-like disease (PMLD) or hypomyelinating leukodystrophy (OMIM number 608804)5 and spastic paraplegia, type 44 (OMIM number 613206)5 in addition to a type of hereditary lymphoedema. Several autosomal recessive pathogenic mutations have been reported in GJC2 gene in association with these disorders6,7,8.
Table.
Summary of variant detection and filtering of the exome sequencing data.

Figure B.
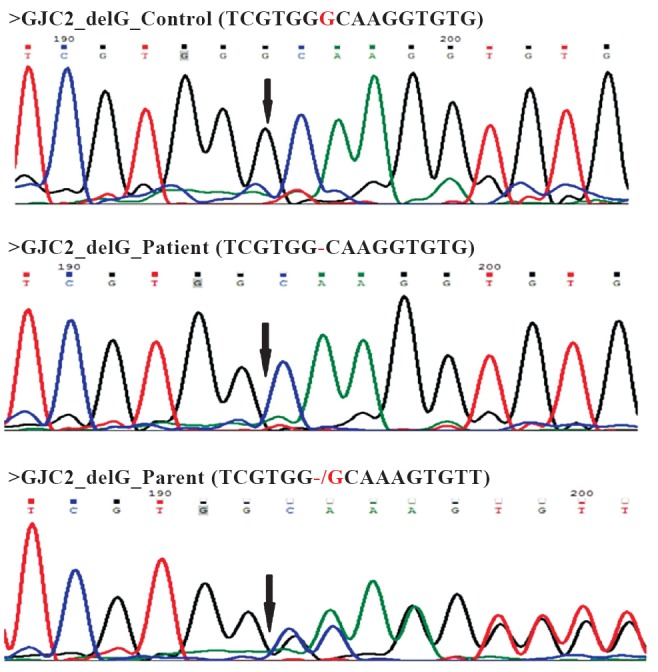
Representative chromatograms of Sanger sequencing showing no deletion in the control, homozygous deletion in the patient A and heterozygous deletion in parent (arrow).
In our case, both the patients were similarly affected with pendular nystagmus, slowly progressive ataxia, later evolving to spastic ataxia with prominent pyramidal signs and eventually spastic paraparesis which is compatible with PMLD. MRI showing changes of hypomyelination or dysmyelination is consistent with the diagnosis. We used a combination of classical homozygosity mapping strategy using microarray data and high throughput exome sequencing to explore the new mutation. In-silico analysis revealed that the deletion of G causes a frameshift at 23rd amino acid creating a stop codon (TGA) at codon number 38 which is likely to result in a truncated protein and thus would likely to be pathogenic in the patients9.
The present study demonstrated the clinical utility of using whole exome sequencing in combination with microarray-based homozygosity mapping approach to identify a molecular cause in two patients from a family with slowly progressive ataxia and nystagmus. Hereditary spastic ataxia is a genetically heterogenous condition and many different genes as well as loci have been identified for this group of ataxias. It is very difficult to narrow down the diagnosis based on clinical features and imaging studies. Genetic analysis for multiple genes is time consuming, tedious and costly. Genetic testing for panel of genes can be done using massively parallel sequencing but it will not identify mutations in hitherto unknown genes for spastic ataxia. Hence, we used array CGH based homozygosity mapping followed by exome sequencing strategy in this family.
Common LOH regions obtained in array CGH based homozygosity mapping were helpful to narrow down the region of interest for variant identification by exome sequencing. This combined approach can act as a rapid and powerful tool in molecular diagnosis of Mendelian disorders for identifying disease causing mutations in autosomal recessive disorders, especially in consanguineous families. Viewed in the context of previous reports10,11, it can be suggested that this combination approach may be considered as a diagnostic option in recessive disorders for which clinical and molecular diagnoses are challenging, especially those for which clinical management may be affected.
Acknowledgment
Authors thank the patients and the family members for their cooperation and Indian Council of Medical Research, New Delhi, and Centre for DNA Fingerprinting and Diagnostics, Hyderabad, for financial support.
Footnotes
Conflicts of Interest: None.
References
- 1.White DR, Ganesh A, Nishimura D, Rattenberry E, Ahmed S, Smith UM, et al. Autozygosity mapping of Bardet-Biedl syndrome to 12q21.2 and confirmation of FLJ23560 as BBS10. Eur J Hum Genet. 2007;15:173–8. doi: 10.1038/sj.ejhg.5201736. [DOI] [PubMed] [Google Scholar]
- 2.Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–3. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bethesda, MD: National Heart, Lung and Blood Institute (NHLBI) (updated July 3, 2013); [accessed on August 18, 2013]. SeattleSeq Annotation 137. Available from: http://snp.gs.washington.edu/SeattleSeqAnnotation137/ [Google Scholar]
- 5.Online Mendelian Inheritance in Man, OMIM. McKusick- Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD), National Human Genome Institute (updated July 3, 2013) [accessed on Augustt 18, 2013]. Available from: http://www.omim.org/
- 6.Orthmann-Murphy JL, Salsano E, Abrams CK, Bizzi A, Uziel G, Freidin MM, et al. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain. 2009;132:426–38. doi: 10.1093/brain/awn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osaka H, Hamanoue H, Yamamoto R, Nezu A, Sasaki M, Saitsu H, et al. Disrupted SOX10 regulation of GJC2 transcription causes Pelizaeus-Merzbacher-like disease. Ann Neurol. 2010;68:250–4. doi: 10.1002/ana.22022. [DOI] [PubMed] [Google Scholar]
- 8.Al-Yahyaee SA, Al-Kindi M, Jonghe PD, Al-Asmi A, Al-Futaisi A, Vriendt ED, et al. Pelizaeus-Merzbacher-like disease in a family with variable phenotype and a novel splicing GJC2 mutation. J Child Neurol. 2013;28:1467–73. doi: 10.1177/0883073812463610. [DOI] [PubMed] [Google Scholar]
- 9.Uhlenberg B, Schuelke M, Ruschendorf F, Ruf N, Kaindl AM, Henneke M, et al. Mutation in the gene encoding gap junction protection alpha-12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. J Hum Genet. 2004;75:251–60. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walsh T, Shahin H, Elkan-Miller T, Lee MK, Thornton AM, Roeb W, et al. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am J Hum Genet. 2010;87:90–4. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cullinane AR, Vilboux T, O’Brien K, Curry JA, Maynard DM, Carlson-Donohoe H, et al. Homozygosity mapping and whole-exome sequencing to detect SLC45A2 and G6PC3 mutations in a single patient with oculocutaneous albinism and neutropenia. J Invest Dermatol. 2011;131:2017–25. doi: 10.1038/jid.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]