

Phospholipids in the plasma membrane of eukaryotic cells are asymmetrically distributed between the outer and inner leaflets.1 Phosphatidylcholine and sphingomyelin are located primarily in the outer leaflet, while phosphatidylserine (PtdSer) and phosphatidylethanolamine (PtdEtn) are restricted to the cytoplasmic side. This asymmetrical phospholipid distribution is disrupted during various biological processes. PtdSer exposed on the outside of activated platelets promotes blood clotting, and on apoptotic cells acts as an “eat me” signal for macrophages. The phospholipid distribution is regulated by lipid transporters in the plasma membrane.1 Scramblases non-specifically and bidirectionally transport phospholipids between the inner and outer leaflets in an ATP-independent manner, while flippases, also known as ATP-dependent aminophospholipid translocases, transport PtdSer and PtdEtn from the extracellular to the cytoplasmic side.1
We recently found that 2 membrane protein families, TMEM16 and XKR, promote phospholipid scrambling.2,3 The TMEM16 family, which has 10 members in humans, contains 8 transmembrane (TM) regions. TMEM16A and 16B act as Ca2+-dependent Cl− channels, while TMEM16C, 16D, 16F, 16G, and 16J promote phospholipid scrambling at plasma membranes in a Ca2+-dependent manner.4 In particular, TMEM16F is responsible for the PtdSer exposure on activated platelets, and Scott syndrome, a bleeding disorder, is caused by mutation in the TMEM16F gene.3 The XKR family is predicted to contain 6 TM regions. Among its 9 human members, XKR8 and some others carry a recognition sequence for caspases 3 and 72 at their C-terminus, and are activated by caspase cleavage to promote phospholipid scrambling.
To identify genes for flippases, we used a genetic screen with the human near-haploid cell line, KBM7. KBM7 cells were mutagenized by retrovirus infection, and a cell population that lost the ability to internalize fluorescently labeled PtdSer was obtained by repeated FACS sorting. The sequencing of millions of mutated alleles in this population led to the identification of ATP11C, a P4-ATPase, and its common β-subunit, CDC50A.5 ATP11C-deficient KBM7 cells and mouse WR19L cells exhibit reduced flippase activity for PtdSer and PtdEtn, while CDC50A-null cells lose the flippase activity almost completely. Thus, CDC50A−/- cells, but not ATP11C−/− cells, constitutively expose PtdSer. Most, if not all, P4-ATPases require CDC50 family proteins as a functional subunit or as a chaperone for plasma membrane localization. Of the 3 CDC50 proteins, KBM7 and WR19L cells express only CDC50A. In contrast, these cells express several P4-ATPases including ATP11C, some of which may have weak flippase activity at the plasma membrane.
Like other P-type ATPases, ATP11C contains 10 TM regions, and a large cytoplasmic region between TM4 and TM5, containing actuator, phosphorylation, and nucleotide-binding domains. ATP11C carries 3 caspase-recognition sites in its nucleotide-binding domain. Caspase-resistant mutation in these 3 sites does not affect ATP11C's flippase activity, but prevents apoptotic cells from exposing PtdSer for their engulfment by macrophages.6 These results indicate that in addition to the caspase-mediated activation of scramblase (Xkr8), the caspase-mediated inactivation of flippase (ATP11C) is required for apoptotic PtdSer exposure. Notably, neither flippase inactivation nor scramblase activation alone is sufficient to present an “eat me” signal on apoptotic cells. Once phospholipid asymmetry is established in plasma membranes, disrupting it without enzymatic help takes a long time (t1/2 of several days).6 Thus, apoptotic cells activate the scramblase to quickly expose PtdSer for efficient engulfment by macrophages. The caspase-mediated apoptotic PtdSer exposure is irreversible, while the PtdSer exposure in activated platelets appears to be reversible. Since P4-type ATPases are inhibited by high Ca2+ concentrations, the TMEM16F-mediated PtdSer exposure in activated platelets is likely to be coupled to the Ca2+-dependent down-regulation of flippases. Once the cellular Ca2+ levels are reduced again, ATP11C would re-establish the asymmetric phospholipid distribution in the plasma membrane.
Since cells expressing a constitutively active TMEM16F mutant (D430G-L) expose PtdSer3 on their surface but are not engulfed by macrophages,7 we previously concluded that PtdSer is necessary but not sufficient to act as an “eat me” signal. However, PtdSer-exposing CDC50A−/− cells are efficiently engulfed by macrophages. Notably, unlike CDC50A−/− cells, the D430G-L cells bind to macrophages at 4°C, but not at 25°C, suggesting that the macrophages’ affinity for the PtdSer exposed on these cells is lower than that for the PtdSer on CDC50A−/− cells. In D430G-L cells, the flippase activity is intact, and PtdSer appears to move between the inner and outer leaflets of the plasma membrane, which could destabilize the interaction between PtdSer and its receptor on macrophages. However, in CDC50A−/− cells, since the flippase is absent, PtdSer exposed to the cell surface will not return to the inner leaflets, so it binds tightly to its receptor on macrophages. These results indicate that once PtdSer is irreversibly exposed on the cell surface, it is sufficient to act as an “eat me” signal, and thus explain why apoptotic cells but not activated platelets are engulfed by macrophages. CDC50A−/− cells grow normally in vitro, suggesting that disrupting the asymmetrical phospholipid distribution does not adversely affect cell growth. However, these cells are cleared in vivo, probably because macrophages engulf them.
In conclusion, flippases and scramblases, the long sought-after molecules involved in the lipid movement at plasma membranes, have been identified in mammals. Studies on the molecular architecture and functional mechanisms of ATP11C, TMEM16F, and Xkr8 will elucidate how phospholipids move between the outer and inner leaflets of plasma membranes.
Figure 1.
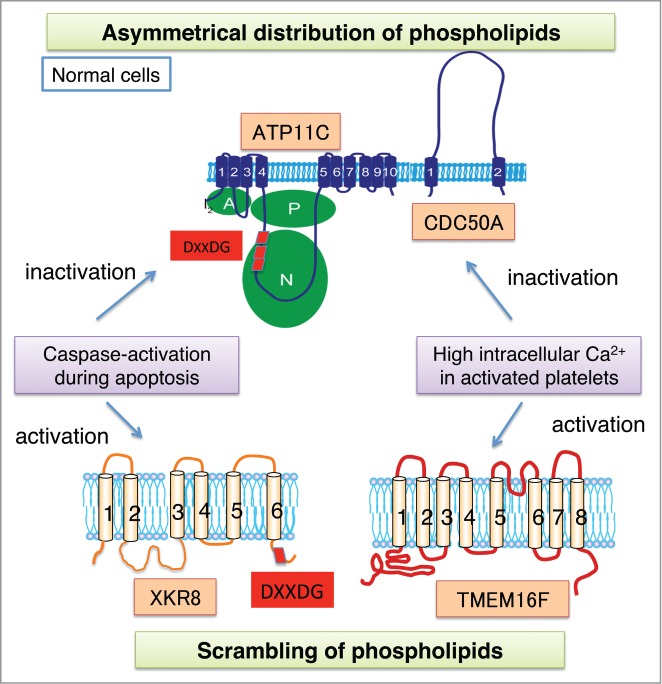
Phospholipids at plasma membranes. In normal cells, phospholipids are asymmetrically distributed between inner and outer leaflets by the function of flippase (ATP11C). During apoptosis, caspases cleave ATP11C to inactivate it, while activating Xkr8 to scramble phospholipids, thus exposing phosphatidylserine to the cell surface. In platelet activation, a high intracellular Ca2+ appears to inactivate ATP11C, while it activates TMEM16-supported phospholipid scrambling.
References
- 1. Leventis PA, et al. . Annu Rev Biophys 2010; 39:407-27; PMID:20192774; http://dx.doi.org/ 10.1146/annurev.biophys.093008.131234 [DOI] [PubMed] [Google Scholar]
- 2. Suzuki J, et al. . Science 2013; 341:403-6; PMID:23845944; http://dx.doi.org/ 10.1126/science.1236758 [DOI] [PubMed] [Google Scholar]
- 3. Suzuki J, et al. . Nature 2010; 468:834-8; PMID:21107324; http://dx.doi.org/ 10.1038/nature09583 [DOI] [PubMed] [Google Scholar]
- 4. Suzuki J, et al. . J Biol Chem 2013; 288:13305-16; PMID:23532839; http://dx.doi.org/ 10.1074/jbc.M113.457937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Segawa K, et al. . Science 2014; 344:1164-8; PMID:24904167; http://dx.doi.org/ 10.1126/science.1252809 [DOI] [PubMed] [Google Scholar]
- 6. Zachowski A. Biochem J 1993; 294:1-14.; PMID:8363559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Segawa K, et al. . Proc Natl Acad Sci U S A 2011; 108:19246-51; PMID:22084121; http://dx.doi.org/ 10.1073/pnas.1114799108 [DOI] [PMC free article] [PubMed] [Google Scholar]