

Update to: European Journal of Human Genetics (2012) 20; doi:10.1038/ejhg.2012.70; published online 18 April 2012
1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Nemaline myopathy (NEM1—NEM10).
Includes nemaline myopathy with excess thin filaments/actin aggregates; nemaline myopathy with cores; nemaline myopathy with intranuclear rods; and Amish nemaline myopathy.
1.2 OMIM# of the disease
NEM1 - 609284; NEM2 - 256030; NEM3 - 161800; NEM4 - 609285; NEM5 - 605355; NEM6 - 609273; NEM7 - 610687; NEM8 - 615348; NEM9 - 615731; NEM10 - 616165.
1.3 Name of the analysed genes or DNA/chromosome segments
Slow muscle α-tropomyosin (TPM3) - NEM1.
Nebulin (NEB) - NEM2.
Skeletal muscle α-actin (ACTA1) - NEM3.
β-tropomyosin (TPM2) - NEM4.
Slow muscle troponin-T (TNNT1) - NEM5.
Kelch-repeat and BTB (POZ) Domain containing 13 (KBTBD13) - NEM6.
Skeletal muscle cofilin (CFL2) - NEM7.
KELCH-like 40 (KLHL40) - NEM8.
KELCH-like 41 (KLHL41) - NEM9.
Leiomodin 3 (LMOD3) - NEM10.
1.4 OMIM# of the gene(s)
TPM3=*191030; NEB=*161650; ACTA1=*102610; TPM2=*190990; TNNT1=*191041; KBTBD13=*613727; CFL2=*601443; KLHL40=*615340; KLHL41=*607701; LMOD3=*616112.
1.5 Mutational spectrum
TPM3: mainly dominant, missense variants;1, 2 however, some recessive variants have been described.3, 4 A 1 bp recessive deletion occurs as a founder variant in the Turkish population.5
NEB: all of the over 140 variants identified to date are recessive and the patients are usually compound heterozygous. The majority of the variants are either frameshift or nonsense variants, but also missense variants, and point variants and deletions affecting splice sites are known.6, 7 An in-frame deletion of exon 55 is present in the Ashkenazi Jewish population at a carrier frequency of ~1 in 108.8 Some patients present with a distal myopathy, with their skeletal muscle biopsies containing nemaline bodies, both nemaline bodies and cores, or no nemaline bodies.9, 10 Rare cases of core-rod myopathy with generalised muscle weakness may also be caused by NEB variants.11
ACTA1: over 200 different variants identified, with the majority causing nemaline myopathy, or nemaline myopathy with other features (eg cores, actin aggregates, intranuclear rods and zebra bodies).12 Of these, most variants are dominant, missense, and have arisen de novo.13 About 10% are recessive variants. Most recessive variants are genetic or functional null variants13 but recently recessive ACTA1 disease with retention of skeletal muscle actin expression was described in a family presenting with a rigid spine syndrome.14 Dominant inheritance is less common, and only seen in families with a milder phenotype.13 Somatic mosaicism is possible with dominant variants.15 Some variants can cause hypercontractile skeletal muscle rather than weakness.16
TPM2: Two heterozygous, dominant missense variants causing nemaline myopathy are known.17 Also a homozygous null variant in a patient with nemaline and Escobar syndrome,18 and a dominant heterozygous variant in a mother with nemaline myopathy and her daughter with cap myopathy19 have been identified. A K7del variant was identified in a family with nemaline bodies and minicores (presenting as a distal myopathy), and also in four unrelated families with distal arthrogryposis type 7 with nemaline bodies.20 The K7del variant also causes progressive skeletal muscle contractures, most probably because of the hypercontractility caused by the mutant protein in nemaline myopathy and/or core-rod myopathy patients.21
TNNT1: a recessive nonsense founder variant is present in the Old Order Amish population. This produces a characteristic progressive nemaline myopathy with tremors and contractures.22 TNNT1 variants have now been identified outside the Amish population.23
KBTBD13: three dominant missense variants have been identified.24 There was phenotypic variability, with some variant carriers exhibiting only very mild proximal leg weakness on targeted examination.
CFL2: homozygous missense variants have been identified in two families,25, 26 and a homozygous 4 bp deletion identified in another family.27 The severity of the disease is greater in the family with the homozygous null variant.
KLHL40: homozygous or compound heterozygous variants (mainly missense, but also frameshift, splice site and nonsense variants) cause severe autosomal recessive nemaline myopathy with prenatal onset of symptoms, including foetal akinesia or hypokinesia and contractures.28 At birth, the children may have fractures, respiratory failure and severe swallowing difficulties.28
KLHL41: Recessive homozygous or compound heterozygous variants have been reported, with frameshift variants leading to a severe phenotype with neonatal death, and missense variants compatible with survival into late childhood or early adulthood.29
LMOD3: Recessive homozygous and compound heterozygous variants cause severe, usually lethal nemaline myopathy.30
Regularly updated variant databases exist for ACTA1, CFL2, KBTBD13, KLHL40, NEB, TNNT1, TPM2 and TPM3 at the Leiden Muscular Dystrophy pages (http://www.dmd.nl).
1.6 Analytical methods
The main analytical method has been bi-directional Sanger sequencing of the entire coding region of the individual genes. If the family structure is amenable, linkage analysis for NEB may be useful to pre-screen, because of the large size of the gene. Next-generation sequencing now allows for simultaneous analysis of all genes in a patient through whole-exome sequencing. Alternatively sub-exomic sequencing using a panel of selected genes can include all known nemaline myopathy genes.31 It should be noted however that limitations of current high throughput sequencing technologies prevent complete screening of all exons in all genes (eg, regions of genes that have high GC content or are repetitive).
1.7 Analytical validation
Variants should be confirmed by resequencing using a fresh dilution of genomic DNA. Putative variants identified through next-generation sequencing methods should be confirmed by Sanger sequencing. Special care should be taken in interpreting missense variants in the nebulin gene as affecting function.10
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence)
For the most part, the frequency of the disease is unknown. In Finland, the birth incidence has been estimated to be 0.02 per 1000 live births.32 De novo dominant variants in ACTA1 and recessive variants in NEB are the most common causes of NEM.33, 34
1.9 If applicable, prevalence in the ethnic group of investigated person
Recessive founder variants are known to exist in particular genes in specific populations: TNNT1 in the Amish;22 NEB in the Ashkenazi Jewish8 and in the Finnish population.10 ACTA1 in the Pakistani community in England, and in French and Spanish Roma;13 TPM3 in the Turkish population;5 and KLHL40 in Japanese persons.28 These specific cases aside, no clear differences in prevalence rates are known between different ethnic groups.
1.10 Diagnostic setting

Comment: requests for predictive testing are not common because of the early onset of the disease, but may be offered in families with childhood or late-onset forms of the disease.
2. TEST CHARACTERISTICS
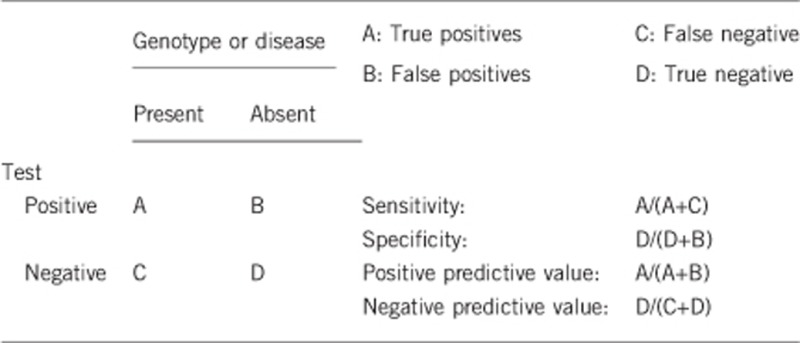
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
100%.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
100%.
2.3 Clinical Sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity is dependent on factors such as age, inheritance pattern and additional clinical features. Because of the genetic heterogeneity, and particularly the difficulty of screening NEB, full screening of all known nemaline myopathy genes has historically not been possible. However, next-generation technologies are now getting closer to such screening. If full screening were to be undertaken, it may be estimated that ~75% of patients would have a variant(s) identified.6, 13
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case. Probably 100%.
2.5 Positive clinical predictive value (life time risk of developing the disease if the test is positive)
Nearly 100%. Potential incomplete penetrance has been suggested for certain ACTA1 variants35 and a very mild phenotype can be associated with some KBTBD13 variants.24
2.6 Negative clinical predictive value (probability of not developing the disease if the test is negative)
Assume an increased risk based on family history for a nonaffected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
Approximately 100%.
Index case in that family had not been tested:
No predictive tests are usually performed in such cases.
3. CLINICAL UTILITY
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
Nemaline myopathy is both a clinical and, significantly, a histopathological/electron microscopic diagnosis. Therefore, a thorough assessment including a detailed evaluation of clinical and pathological features should be performed along with genetic testing. As such, histopathology and electron microscopy are not diagnostic alternatives, rather prerequisites to genetic testing. Nevertheless, muscle biopsy is an invasive procedure, and appropriate histological and electron microscopic examination requires proximity to a specialised laboratory set up.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Not applicable.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
Predictive testing is usually only applicable for the milder versions of nemaline myopathy, as most often the disease presents before, at or shortly after birth.
3.2.1 Will the result of a genetic test influence lifestyle and prevention?

3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
Not applicable.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes, if a variant/s is identified.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Genetic testing would still most likely be performed for other family members, however, other tests such as skeletal muscle biopsy might be prevented.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes, but owing to very early disease onset in most cases, it is infrequently encountered.
3.4 Prenatal diagnosis
(To be answered if in 1.10 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes. In cases where dominant de novo variants have been identified in an affected child, genetic counselling may be difficult, as the recurrence risk is between 0 and 25% because of the possibility of gonadal mosaicism. However, prenatal genetic testing of an at-risk pregnancy is accurate.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe).
Yes, particularly if a variant/s are identified. An accurate genetic diagnosis often ends a lengthy diagnostic odyssey for the patient and their family, removing the psychological effects of an absent disease, and can sometimes influence possible prognosis. An accurate genetic diagnosis can crucially indicate the mode of inheritance and underpins genetic counselling, including options such as prenatal and pre-implantation testing.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469), the European Society of Human Genetics, an Australian National Health and Medical Research Council (NH&MRC) Fellowship APP1002147 and an Australian Research Council (ARC) Future Fellowship FT100100734.
The authors declare no conflict of interest.
References
- Laing NG, Wilton SD, Akkari PA et al: A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy. Nat Genet 1995; 9: 75–79. [DOI] [PubMed] [Google Scholar]
- Kiphuth IC, Krause S, Huttner HB, Dekomien G, Struffert T, Schroder R: Autosomal dominant nemaline myopathy caused by a novel alpha-tropomyosin 3 mutation. J Neurol 2010; 257: 658–660. [DOI] [PubMed] [Google Scholar]
- Tan P, Briner J, Boltshauser E et al: Homozygosity for a nonsense mutation in the alpha-tropomyosin slow gene TPM3 in a patient with severe infantile nemaline myopathy. Neuromuscul Disord 1999; 9: 573–579. [DOI] [PubMed] [Google Scholar]
- Wattanasirichaigoon D, Swoboda KJ, Takada F et al: Mutations of the slow muscle alpha-tropomyosin gene, TPM3, are a rare cause of nemaline myopathy. Neurology 2002; 59: 613–617. [DOI] [PubMed] [Google Scholar]
- Lehtokari VL, Pelin K, Donner K et al: Identification of a founder mutation in TPM3 in nemaline myopathy patients of Turkish origin. Eur J Hum Genet 2008; 16: 1055–1061. [DOI] [PubMed] [Google Scholar]
- Pelin K, Hilpela P, Donner K et al: Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci USA 1999; 96: 2305–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtokari VL, Pelin K, Sandbacka M et al: Identification of 45 novel mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Hum Mutat 2006; 27: 946–956. [DOI] [PubMed] [Google Scholar]
- Anderson SL, Ekstein J, Donnelly MC et al: Nemaline myopathy in the Ashkenazi Jewish population is caused by a deletion in the nebulin gene. Hum Genet 2004; 115: 185–190. [DOI] [PubMed] [Google Scholar]
- Scoto M, Cullup T, Cirak S et al: Nebulin (NEB mutations in a childhood onset distal myopathy with rods and cores uncovered by next generation sequencing. Eur J Hum Genet 2013; 21: 1249–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtokari VL, Kiiski K, Sandaradura SA et al: Mutation update: the spectra of nebulin variants and associated myopathies. Hum Mutat 2014; 35: 1418–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero NB, Lehtokari VL, Quijano-Roy S et al: Core-rod myopathy caused by mutations in the nebulin gene. Neurology 2009; 73: 1159–1161. [DOI] [PubMed] [Google Scholar]
- Nowak KJ, Ravenscroft G, Laing NG: Skeletal muscle alpha-actin diseases (actinopathies): pathology and mechanisms. Acta Neuropathologica 2013; 125: 12–29. [DOI] [PubMed] [Google Scholar]
- Laing NG, Dye DE, Wallgren-Pettersson C et al: Mutations and polymorphisms of the skeletal muscle alpha-actin gene (ACTA1. Hum Mutat 2009; 30: 1267–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Grady GL, Best HA, Oates EC et al: Recessive ACTA1 variant causes congenital muscular dystrophy with rigid spine. Eur J Hum Genet 2014, e-pub ahead of print 3 September 2014; doi:10.1038/ejhg.2014.169. [DOI] [PMC free article] [PubMed]
- Nowak KJ, Wattanasirichaigoon D, Goebel HH et al: Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet 1999; 23: 208–212. [DOI] [PubMed] [Google Scholar]
- Jain RK, Jayawant S, Squier W et al: Nemaline myopathy with stiffness and hypertonia associated with an ACTA1 mutation. Neurology 2012; 78: 1100–1103. [DOI] [PubMed] [Google Scholar]
- Donner K, Ollikainen M, Ridanpaa M et al: Mutations in the beta-tropomyosin (TPM2 gene–a rare cause of nemaline myopathy. Neuromuscul Disord 2002; 12: 151–158. [DOI] [PubMed] [Google Scholar]
- Monnier N, Lunardi J, Marty I et al: Absence of beta-tropomyosin is a new cause of Escobar syndrome associated with nemaline myopathy. Neuromuscul Disord 2009; 19: 118–123. [DOI] [PubMed] [Google Scholar]
- Tajsharghi H, Ohlsson M, Lindberg C, Oldfors A: Congenital myopathy with nemaline rods and cap structures caused by a mutation in the beta-tropomyosin gene (TPM2. Arch Neurol 2007; 64: 1334–1338. [DOI] [PubMed] [Google Scholar]
- Davidson AE, Siddiqui FM, Lopez MA et al: Novel deletion of lysine 7 expands the clinical, histopathological and genetic spectrum of TPM2-related myopathies. Brain 2013; 136: 508–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokbel N, Ilkovski B, Kreissl M et al: K7del is a common TPM2 gene mutation associated with nemaline myopathy and raised myofibre calcium sensitivity. Brain 2013; 136: 494–507. [DOI] [PubMed] [Google Scholar]
- Johnston JJ, Kelley RI, Crawford TO et al: A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am J Hum Genet 2000; 67: 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Pol WL, Leijenaar JF, Spliet WG et al: Nemaline myopathy caused by TNNT1 mutations in a Dutch pedigree. Mol Genet Genomic Med 2014; 2: 134–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambuughin N, Yau KS, Olive M et al: Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. Am J Hum Genet 87: 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal PB, Greenleaf RS, Tomczak KK et al: Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am J Hum Genet 2007; 80: 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ockeloen CW, Gilhuis HJ, Pfundt R et al: Congenital myopathy caused by a novel missense mutation in the CFL2 gene. Neuromuscul Disord 2012; 22: 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong RW, Alsaman A, Selcen D et al: Novel cofilin-2 (CFL2 four base pair deletion causing nemaline myopathy. J Neurol Neurosurg Psychiatry 2014; 85: 1058–1060. [DOI] [PubMed] [Google Scholar]
- Ravenscroft G, Miyatake S, Lehtokari VL et al: Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am J Hum Genet 2013; 93: 6–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta VA, Ravenscroft G, Shaheen R et al: Identification of KLHL41 mutations implicates BTB-Kelch-mediated ubiquitination as an alternate pathway to myofibrillar disruption in nemaline myopathy. Am J Hum Genet 2013; 93: 1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen M, Sandaradura SA, Dowling JJ et al: Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest 2014; 124: 4693–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiiski K, Laari L, Lehtokari VL et al: Targeted array comparative genomic hybridization-a new diagnostic tool for the detection of large copy number variations in nemaline myopathy-causing genes. Neuromuscul Disord 2013; 23: 56–65. [DOI] [PubMed] [Google Scholar]
- Wallgren-Pettersson C: Congenital nemaline myopathy: a longitudinal study. University of Helsinki: Helsinki, 1990. [Google Scholar]
- Wallgren-Pettersson C, Pelin K, Hilpela P et al: Clinical and genetic heterogeneity in autosomal recessive nemaline myopathy. Neuromuscul Disord 1999; 9: 564–572. [DOI] [PubMed] [Google Scholar]
- Wallgren-Pettersson C, Laing NG: Report of the 83rd ENMC International Workshop: 4th Workshop on Nemaline Myopathy, 22-24 September 2000, Naarden, The Netherlands. Neuromuscul Disord 2001; 11: 589–595. [DOI] [PubMed] [Google Scholar]
- Agrawal PB, Strickland CD, Midgett C et al: Heterogeneity of nemaline myopathy cases with skeletal muscle alpha-actin gene mutations. Ann Neurol 2004; 56: 86–96. [DOI] [PubMed] [Google Scholar]
- Jungbluth H, Sewry CA, Brown SC et al: Mild phenotype of nemaline myopathy with sleep hypoventilation due to a mutation in the skeletal muscle alpha-actin (ACTA1 gene. Neuromuscul Disord 2001; 11: 35–40. [DOI] [PubMed] [Google Scholar]
- Hutchinson DO, Charlton A, Laing NG, Ilkovski B, North KN: Autosomal dominant nemaline myopathy with intranuclear rods due to mutation of the skeletal muscle ACTA1 gene: clinical and pathological variability within a kindred. Neuromuscul Disord 2006; 16: 113–121. [DOI] [PubMed] [Google Scholar]
- Laing NG, Clarke NF, Dye DE et al: Actin mutations are one cause of congenital fibre type disproportion. Ann Neurol 2004; 56: 689–694. [DOI] [PubMed] [Google Scholar]
- Clarke NF, Kolski H, Dye DE et al: Mutations in TPM3 are a common cause of congenital fiber type disproportion. Ann Neurol 2008; 63: 329–337. [DOI] [PubMed] [Google Scholar]
- Lawlor MW, Dechene ET, Roumm E, Geggel AS, Moghadaszadeh B, Beggs AH: Mutations of tropomyosin 3 (TPM3 are common and associated with type 1 myofiber hypotrophy in congenital fiber type disproportion. Hum Mutat 2010; 31: 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung RM, Yoon G, Hawkins CE, Halliday W, Biggar D, Vajsar J: Cap myopathy caused by a mutation of the skeletal alpha-actin gene ACTA1. Neuromuscul Disord 2010; 20: 238–240. [DOI] [PubMed] [Google Scholar]
- Lehtokari VL, Ceuterick-de Groote C, de Jonghe P et al: Cap disease caused by heterozygous deletion of the beta-tropomyosin gene TPM2. Neuromuscul Disord 2007; 17: 433–442. [DOI] [PubMed] [Google Scholar]
- Clarke NF, Domazetovska A, Waddell L, Kornberg A, McLean C, North KN: Cap disease due to mutation of the beta-tropomyosin gene (TPM2. Neuromuscul Disord 2009; 19: 348–351. [DOI] [PubMed] [Google Scholar]
- Ohlsson M, Fidzianska A, Tajsharghi H, Oldfors A: TPM3 mutation in one of the original cases of cap disease. Neurology 2009; 72: 1961–1963. [DOI] [PubMed] [Google Scholar]
- De Paula AM, Franques J, Fernandez C et al: A TPM3 mutation causing cap myopathy. Neuromuscul Disord 2009; 19: 685–688. [DOI] [PubMed] [Google Scholar]
- Wallgren-Pettersson C, Lehtokari VL, Kalimo H et al: Distal myopathy caused by homozygous missense mutations in the nebulin gene. Brain 2007; 130: 1465–1476. [DOI] [PubMed] [Google Scholar]
- Lehtokari VL, Pelin K, Herczegfalvi A et al: Nemaline myopathy caused by mutations in the nebulin gene may present as a distal myopathy. Neuromuscul Disord 2011; 21: 556–562. [DOI] [PubMed] [Google Scholar]
- Sung SS, Brassington AM, Grannatt K et al: Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am J Hum Genet 2003; 72: 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scacheri PC, Hoffman EP, Fratkin JD et al: A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology 2000; 55: 1689–1696. [DOI] [PubMed] [Google Scholar]
- Clarke NF, Waddell LB, Cooper ST et al: Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum Mutat 2010; 31: E1544–E1550. [DOI] [PubMed] [Google Scholar]
- Clarke NF, Kidson W, Quijano-Roy S et al: SEPN1: associated with congenital fiber-type disproportion and insulin resistance. Ann Neurol 2006; 59: 546–552. [DOI] [PubMed] [Google Scholar]
- Munot P, Lashley D, Jungbluth H et al: Congenital fibre type disproportion associated with mutations in the tropomyosin 3 (TPM3 gene mimicking congenital myasthenia. Neuromuscul Disord 2010; 20: 796–800. [DOI] [PubMed] [Google Scholar]
- Rodriguez Cruz PM, Sewry C, Beeson D et al: Congenital myopathies with secondary neuromuscular transmission defects; a case report and review of the literature. Neuromuscul Disord 2014; 24: 1103–1110. [DOI] [PubMed] [Google Scholar]
- Bethlem J, Arts WF, Dingemans KP: Common origin of rods, cores, miniature cores, and focal loss of cross-striations. Arch Neurol 1978; 35: 555–566. [DOI] [PubMed] [Google Scholar]
- Feinberg DM, Spiro AJ, Weidenheim KM: Distinct light microscopic changes in human immunodeficiency virus-associated nemaline myopathy. Neurology 1998; 50: 529–531. [DOI] [PubMed] [Google Scholar]
- Wallgren-Pettersson C, Laing NG: Report of the 70th ENMC International Workshop: nemaline myopathy, 11–13 June 1999, Naarden, The Netherlands. Neuromuscul Disord 2000; 10: 299–306. [DOI] [PubMed] [Google Scholar]
- D'Amico A, Graziano C, Pacileo G et al: Fatal hypertrophic cardiomyopathy and nemaline myopathy associated with ACTA1 K336E mutation. Neuromuscul Disord 2006; 16: 548–552. [DOI] [PubMed] [Google Scholar]