

Update to: European Journal of Human Genetics (2012) 20; doi:10.1038/ejhg.2012.62; published online 18 April 2012
1. Disease characteristics
1.1 Name of the disease (synonyms)
Spinal muscular atrophy (SMA) type I–IV,1 SMA 5q, proximal SMA, infantile SMA, Werdnig–Hoffmann disease (SMA I), intermediate SMA (SMA II), juvenile spinal muscular atrophy type Kugelberg–Welander (SMA III) and adult onset SMA (SMA IV).
1.2 OMIM# of the disease
253300 (SMA I), 253550 (SMA II), 253400 (SMA III) and 271150 (SMA IV).
1.3 Name of the analyzed genes or DNA/chromosome segments
SMN1 and SMN22/5q11.2-13.3.
1.4 OMIM# of the gene(s)
600354 (SMN1)/601627 (SMN2).
1.5 Mutational spectrum
Comment: The following information is limited to variants of the SMN1 gene. SMN2 gene copy number varies in the normal population from 0 to 3, with ~5–10% of normal individuals having no SMN2 copy. In the presence of at least one SMN1 copy, SMN2 copies do not contribute much to protein expression. Since a homozygous loss of both SMN genes is believed to result in embryonic lethality, the presence of a SMN2 deletion excludes SMA 5q based on a homozygous SMN1 loss of function mutation. Genetic variants of SMN1 include whole-gene deletions, single exon deletions, point mutations, genomic rearrangements (http://www.hgmd.cf.ac.uk/ac/index.php), see also databases for muscular dystrophy and motor neuron diseases including SMN1 mutations (http://grenada.lumc.nl/LSDB_list/lsdbs/SMN1 and http://www.dmd.nl/nmdb2/home.php?select_db=SMN1). For polymorphisms see NCBI accession number NM_000344.3. For SNPs see http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?geneId=6606. For frequency of genotypes in patients see 2.3.
1.6 Analytical methods
PCR (restriction digest).3 Competitive PCR,4 real-time PCR on lightCycler5 or TaqMan basis.6 Microarray typing for detection of large deletions. Multiplex ligation-dependent probe amplification (MLPA).7 Sanger sequencing of the total coding region and the exon–intron boundaries of the SMN1 gene. If an intragenic variant is detected, it is necessary to verify that the variant has occurred in SMN1 and not SMN2. This requires additional testing by a method that facilitates SMN1-specific and SMN2-specific amplification and sequence analysis, e.g., by long-range PCR protocol8 or subcloning.9, 10
1.7 Analytical validation
All mutations identified should be confirmed by a second, independent test (PCR, quantitative PCR, sequencing and MLPA). It is recommended to confirm the segregation of the mutation in the parents. For intragenic missense mutations to be considered pathogenic, they should not be described in the literature in control alleles, should be in evolutionary conserved regions and should be predicted by applicable software to be probably pathogenic. If feasible, patient tissue might be investigated for SMN protein staining.
1.8 Estimated frequency of the disease (Incidence at birth (‘birth prevalence') or population prevalence)
Birth prevalence among Caucasians: 1:10.00011
1.9 If applicable, prevalence in the ethnic group of investigated person
Birth prevalence is much higher in certain inbred populations.
1.10 Diagnostic setting

Comment:
Predictive testing in SMA should be considered on an individual case basis only, as long as no preventive treatment is available. Children at risk should not be tested according to the European guidelines for genetic testing in minors. In addition, it has to be offered with caution, since a small proportion of subjects with homozygous SMN1 deletions/mutations will not develop the disease.
2. Test characteristics
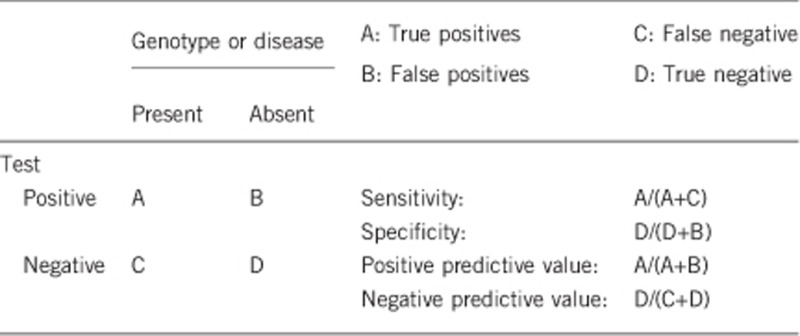
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Depending on the ethnic origin and the applied methods:
PCR, restriction digest: >90% (only homozygous SMN1 exon 7 and 8 deletions)
MLPA/quantitative PCR: 100% (homozygous SMN1 deletion), heterozygous SMN1 deletion
MLPA/quantitative PCR plus SMN1 sequencing: >99% (compound heterozygous SMN1 deletion/mutation)
Comment: Screening does not detect the extremely rare homozygous point mutations.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
PCR, restriction digest: >90%
MLPA/quantitative PCR: >99% for homozygous/heterozygous SMN1 (and SMN2) deletions, no information about rare point mutations
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present) Proportions of genotypes among patients with mutations of the SMN1 gene (SMA 5q):
SMN1 homozygous deletion exon 7 and 8: 85–90%
SMN1 homozygous deletion exon 7 only: 5–10%
SMN1 homozygous deletion exon 8 only: exceptionally12
SMN1 compound heterozygous deletion exon 7/8 and subtle mutation (intragenic deletion or duplication or point mutation): 2–5%10, 13
SMN1 homozygous subtle mutation: exceptionally, restricted to consanguineous families14
Proportion of patients with the clinical picture of proximal SMA displaying mutations of the SMN1 gene:
The sensitivity for SMN1 deletion and sequence analysis for the detection of SMA types I–III is probably almost 100% in a clinically well-defined patient group. Those patients with the clinical picture of SMA I–III not showing SMN1 mutations (non 5q-SMA) represent other genetic and non-genetic entities. The proportion of non 5q-SMA is <2% in SMA I+II, <10% in SMA III, while only few (<10%) of SMA IV patients are caused by SMN1 mutations.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
>99% for SMN1 homozygous deletion/mutation.
90–95% for SMN2 homozygous deletion.
2.5 Positive clinical predictive value
(life time risk to develop the disease if the test is positive)
>99% for homozygous SMN1 deletions, compound heterozygous SMN1 deletion/subtle mutation or homozygous subtle mutations. A small fraction (<1%) of individuals with homozygous SMN1 deletion/mutation will not develop clinical features.
0% for homozygous SMN2 deletions (according to current knowledge, see 1.5)
2.6 Negative clinical predictive value
(Probability not to develop the disease if the test is negative)
Index case in that family had been tested and was positive for a homozygous SMN1 deletion/mutation:
>99% (for a homozygous SMN1 deletion/mutation)
Index case in that family had not been tested for SMN1 deletion/mutation:
>95% for SMA I+II, >90% for SMA III, <10% for SMA IV (for a homozygous SMN1 deletion/mutation)
3. Clinical utility
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

Comment: electrophysiology (electromyography, electroneurography) and muscle biopsy are generally applicable to diagnose anterior horn cell disease.
3.1.2 Describe the burden of alternative diagnostic methods to the patient
Electrophysiology (electromyography, electroneurography) and muscle biopsy are painful and invasive means and will not specify the underlying genetic defect.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Genetic testing is much cheaper than clinical neurological work-up including electrophysiology and muscle biopsy.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe) No.If the test result is negative (please describe) No.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
None.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes, it confirms the mode of inheritance and is the prerequisite for genetic risk assessment in relatives.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes, in order to clarify the diagnosis and the mode of inheritance. No, as regards genetic risk calculation for unaffected relatives.
Proportion of heterozygous carriers for infantile SMA displaying mutations of the SMN1 gene:
Carrier rates are highest in Caucasian populations (1 in 47) and lowest in African Americans (1 in 72) with a pan-ethnic carrier frequency of 1 in 54.15 The test sensitivity for heterozygous carriers does not exceed 92–97% because two or more SMN1 copies are present on about 3–8% of normal chromosomes in most populations apart from African Americans.15 Alleles containing more than one SMN1 gene copy mask a deletion of SMN1 on the other allele. The frequency of these 2-copy SMN1 alleles varies significantly by ethnicity and is highest in African Americans (27%) and lowest (3–4%) in Caucasians.15 The risk reduction of a quantitative carrier screening test also depends on the gene frequency in the ethnic group. Altogether all but African Americans have a carrier detection rate exceeding 90%.15 If a parent shows two SMN1 copies in the quantitative test, further segregation tests including the grandparents are feasible to determine whether the deletion occurred as a de novo event.
New mutations in SMA occur with a frequency of 0.84% of chromosomes,11, 16 and subtle SMN1 gene mutations have been observed in about 2–5% of patients, that is, ~2% of disease alleles.10, 13 Given the low incidence of subtle mutations in the general population, it is not advised to exclude those for carrier-risk estimation unless the family history is positive for such a mutation.
For indications of carrier testing, SMN2 copy number determination does not provide useful additional information. The SMN2 copy number indicates the total copy number for both alleles, therefore, it is not possible to determine the SMN2 phase in unaffected individuals.17
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes, but it is only exceptionally requested. For limitations see 1.10.
3.4 Prenatal diagnosis
(To be answered if in 1.10 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes. It is advised to confirm SMA carrier status in both parents before prenatal diagnosis.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Yes, genetic testing is the gold standard for confirmation of the diagnosis and the mode of inheritance, helps to avoid unnecessary and invasive diagnostic procedures. It allows prognostic evaluations and is the prerequisite for prenatal testing, preimplantation genetic diagnosis and genetic risk estimation of relatives.
The authors declare no conflict of interest.
References
- Munsat TL, Davies KE: International SMA Consortium Meeting: Meeting report. Neuromusc Disord 1992; 2: 423–428. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S et al: Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995; 80: 155–165. [DOI] [PubMed] [Google Scholar]
- Van der Steege G, Grootscholten PM, van der Vlies P et al: PCR-based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet 1995; 345: 985–986. [PubMed] [Google Scholar]
- McAndrew PE, Parsons DW, Simard LR et al: Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet 1997; 60: 1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldkötter M, Schwarzer V, Wirth R et al: Quantitative analyses of SMN1 and SMN2 based on real-time LightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002; 70: 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anhuf D, Eggermann T, Rudnik-Schöneborn S et al: Determination of SMN1 and SMN2 copy number using TaqManTM technology. Hum Mutat Genet 2003; 22: 74–78. [DOI] [PubMed] [Google Scholar]
- Scarciolla O, Stuppia L, De Angelis MV et al: Spinal muscular atrophy genotyping by gene dosage using multiple ligation-dependent probe amplification. Neurogenetics 2006; 7: 269–276. [DOI] [PubMed] [Google Scholar]
- Clermont O, Burlet P, Benit P et al: Molecular analysis of SMA patients without homozygous SMN1 deletions using a new strategy for identification of SMN1 subtle mutations. Hum Mutat 2004; 24: 417–427. [DOI] [PubMed] [Google Scholar]
- Parsons DW, McAndrew PE, Iannaconne ST et al: Intragenic telSMN mutations: Frequency, distribution, evidence of a founder effect, and modifications of the spinal muscular atrophy phenotype by cenSMN copy number. Am J Hum Genet 1998; 63: 1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B, Herz M, Wetter A et al: Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Med Genet 1999; 64: 1340–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino S, Wilson RB: Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum Genet 2002; 111: 477–500. [DOI] [PubMed] [Google Scholar]
- Gambardella A, Mazzei R, Toscano A et al: Spinal muscular atrophy due to an isolated deletion of exon 8 of the telomeric survival motor neuron gene. Ann Neurol 1998; 44: 836–839. [DOI] [PubMed] [Google Scholar]
- Alías L, Bernal S, Fuentes-Prior P et al: Mutation update of spinal muscular atrophy in Spain: molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum Genet 2009; 125: 29–39. [DOI] [PubMed] [Google Scholar]
- Cuscó I, López E, Soler-Botija C, Jesús Barceló M, Baiget M, Tizzano EF: A genetic and phenotypic analysis in Spanish spinal muscular atrophy patients with c.399_402del AGAG, the most frequently found subtle mutation in the SMN1 gene. Hum Mutat 2003; 22: 136–143. [DOI] [PubMed] [Google Scholar]
- Sugarman EA, Nagan N, Zhu H et al: Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72 400 specimens. Eur J Hum Genet 2012; 20: 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B, Schmidt T, Hahnen E et al: De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet 1997; 61: 1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior TW, Narasimhan N, Sugarman EA, Batish SD, Braastad C: Technical standards and guidelines for spinal muscular atrophy testing. Genet Med 2011; 13: 686–694. [DOI] [PubMed] [Google Scholar]