

Abstract
The Kelch-like ECH associated protein 1 (Keap1)–NF-E2 p45-related factor 2 (Nrf2) pathway regulates networks of proteins that protect against the cumulative damage of oxidants, electrophiles and misfolded proteins. The interaction between transcription factor Nrf2 and its main negative cytoplasmic regulator Keap1 follows a cycle whereby the protein complex sequentially adopts two conformations: ‘open’, in which Nrf2 binds to one monomer of Keap1, followed by ‘closed’, in which Nrf2 interacts with both members of the Keap1 dimer. Electrophiles and oxidants (inducers) are recognized by cysteine sensors within Keap1, disrupting its ability to target Nrf2 for ubiquitination and degradation. Consequently, the protein complex accumulates in the ‘closed’ conformation, free Keap1 is not regenerated and newly synthesized Nrf2 is stabilized to activate target-gene transcription. The prevailing view of the Keap1–Nrf2 pathway, for which there exists a wealth of experimental evidence, is that it lies at the heart of cellular defence, playing crucial roles in adaptation and survival under conditions of stress. More recently, the significance of Nrf2 in intermediary metabolism and mitochondrial physiology has also been recognized, adding another layer of cytoprotection to the repertoire of functions of Nrf2. One way by which Nrf2 influences mitochondrial activity is through increasing the availability of substrates (NADH and FADH2) for respiration. Another way is through accelerating fatty acid oxidation (FAO). These findings reinforce the reciprocal relationship between oxidative phosphorylation and the cellular redox state, and highlight the key role of Nrf2 in regulating this balance.
Keywords: cytoprotection, FRET/FLIM, mitochondria, PINK1, redox
Introduction
The transcription factor NF-E2 p45-related factor 2 (Nrf2, gene name NFE2L2) and its negative regulator Kelch-like ECH associated protein 1 (Keap1) control the expression of large networks of genes with versatile cytoprotective activities. At homoeostatic conditions, Keap1 [1], a substrate adaptor protein for cullin3–RING-box protein 1 ubiquitin ligase, continuously targets Nrf2 for ubiquitination and proteasomal degradation [2–4], but loses this function in response to electrophiles and oxidants (termed inducers), which chemically modify reactive cysteine sensors in Keap1 [5,6]. Consequently, Nrf2 accumulates and activates transcription of its downstream target genes encoding enzymes of xenobiotic metabolism, proteins with anti-inflammatory functions, as well as those which regulate cellular redox homoeostasis and intermediary metabolism [7–9].
The cyclic sequential attachment and regeneration model of Keap1-mediated degradation of Nrf2
The use of a quantitative FRET-based methodology combined with multiphoton Fluorescence Lifetime Imaging Microscopy (FLIM) to study the interaction between Keap1 and Nrf2 in single live cells expressing Keap1–mCherry and EGFP–Nrf2 fusion proteins revealed that there are two distinct FRET efficiency (E-FRET) populations (Figures 1A and 1C) [10]. Previous studies have established that Nrf2 binds to dimeric Keap1 via two motifs located in the N-terminal Nrf2-ECH homology (Neh2) domain of the transcription factor, the ‘DLG’ motif, which interacts with one member of the Keap1 dimer and the ‘ETGE’ motif, which binds to the other member of the Keap1 dimer; importantly, the affinity of Keap1 for the ‘ETGE’ motif is 200-fold greater than for the ‘DLG’ [11,12]. The generation of Nrf2 mutants that bind with either lower or higher affinity to Keap1 than wild-type (WT) Nrf2 showed that if the low affinity ‘DLG’ motif in Nrf2 is mutated, so that Nrf2 is bound to only a single member of the Keap1 dimer, the E-FRET signal at 21% is lost, whereas the E-FRET signal at 13% is maintained (Figure 1D). Conversely, when the ‘DLG’ motif is replaced with a second high affinity ‘ETGE’ motif, there is an increase in the signal at 21% E-FRET and a decrease in the signal at 13% E-FRET (Figure 1E). These data suggest that in the basal state the Keap1–Nrf2 complex is found in two distinct conformations, which we have termed ‘open’ and ‘closed’. In the ‘open’ conformation, representing the 13% E-FRET population, only the high affinity ‘ETGE’ motif of Nrf2 is bound to Keap1. In the ‘closed’ conformation, representing the 21% E-FRET population, both the low affinity ‘DLG’ and the high affinity ‘ETGE’ motifs of Nrf2 are bound to the Keap1 dimer (Figure 1A).
Figure 1. Single-cell analysis of the Keap1-Nrf2 interactions.

(A) The ‘open’ and ‘closed’ conformations of the Keap1–Nrf2 complex. The interactions between Keap1 and Nrf2 are shown schematically above the E-FRET distribution. In the images, Keap1 is shown in blue, mCherry in red, Nrf2 in yellow and EGFP in green. On the left, the Keap1–Nrf2 complex is shown in the ‘open’ conformation, in which only the ‘ETGE’ motif of Nrf2 is bound to the Keap1 dimer. This conformation generates the 13% E-FRET population shown in the graph. On the right, the ‘closed’ conformation is shown, where both the ‘DLG’ and ‘ETGE’ motifs of Nrf2 are bound to the Keap1 dimer. This conformation generates the 21% E-FRET population shown below in the graph. (B–E) Human embryonic kidney (HEK)293 cells were imaged 24 h after co-transfection with: WT EGFP–Nrf2 + mCherry (B), WT EGFP–Nrf2 + Keap1–mCherry (C), EGFP–Nrf2ΔDLG + Keap1–mCherry (D) or EGFP–Nrf2 double ETGE + Keap1–mCherry (E). The left columns show the EGFP image. The middle columns show pictorial representations of the E-FRET, where the colour of the cell corresponds to the E-FRET, ranging from 0% to 30%. The right columns show the E-FRET data from each pixel of the image plotted on a graph. The E-FRET in a cell co-transfected with WT EGFP–Nrf2 + free mCherry (B) is 0, corresponding to the blue colour of the cell. The E-FRET distribution in a cell co-transfected with WT EGFP–Nrf2 + Keap1–mCherry (C) shows two distinct peaks, one centred at 13% and the other at 21%, suggesting that there are two different FRET interactions between the EGFP and mCherry fluorophores within the Keap1–Nrf2 complex. These E-FRET populations are shown pictorially in the middle column of (C), where both green and yellow colours can be seen. The E-FRET distribution in a cell co-transfected with EGFP–Nrf2ΔDLG + Keap1–mCherry (D) shows one major peak centred at 13%, indicating that there is a single interaction between the EGFP and mCherry fluorophores within the Keap1–Nrf2ΔDLG complex. This E-FRET population is shown pictorially in the middle column of (D), where the green colour is distributed evenly across the cell. The E-FRET distribution in a cell co-transfected with EGFP–Nrf2 double ETGE + Keap1–mCherry (E) shows one major peak centred at 21%, indicating that there is one major interaction between the EGFP and mCherry fluorophores within the Keap1–Nrf2 double ETGE complex. This E-FRET population is shown pictorially in the middle column of (E), where the predominant colour is yellow. Scale bar=10 μm.
By blocking protein degradation by the proteasome or protein synthesis globally, both of which lead to a decrease in the abundance of the ‘open’ conformation and a corresponding increase in the ‘closed’ conformation, it was established that the two conformations of the Keap1–Nrf2 complex represent two phases of a cycle (Figure 2A). The first phase of the cycle is the ‘open’ conformation, which is decreased by blocking protein synthesis as new Keap1–Nrf2 complexes cannot be formed. The second phase is the ‘closed’ conformation, which is increased by blocking the proteasome as Nrf2 cannot be degraded. Together, the data led us to conclude that in the basal state, one member of a free Keap1 dimer first binds Nrf2 through its high affinity ‘ETGE’ motif to form the ‘open’ conformation of the Keap1–Nrf2 complex. This is followed by binding of the second monomer of Keap1 to the low affinity ‘DLG’ motif of Nrf2 to form the ‘closed’ conformation. The two-site binding positions the lysine residues in the α-helix between the ‘DLG’ and ‘ETGE’ motifs in the correct orientation for ubiquitination by the Keap1-dependent E3-ligase. Consequently, ubiquitinated Nrf2 is released from Keap1 and degraded by the proteasome and free Keap1 is regenerated allowing the cycle to begin again.
Figure 2. The cyclic sequential attachment and regeneration model of Keap1-mediated degradation of Nrf2.
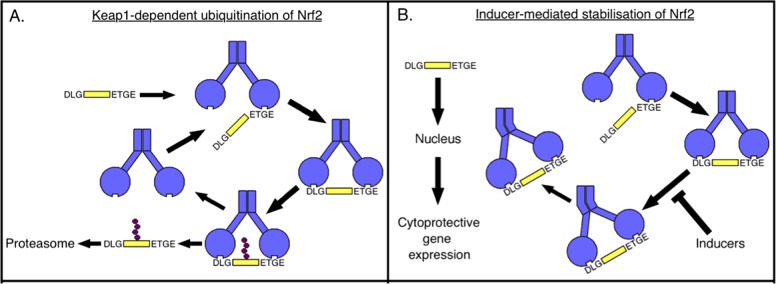
(A) In the basal state, one member of the Keap1 (blue) dimer binds Nrf2 (yellow) first through the high affinity ‘ETGE’ motif to form the ‘open’ conformation of the Keap1–Nrf2 complex. The second monomer of Keap1 then binds to the low affinity ‘DLG’ motif of Nrf2 to form the ‘closed’ conformation. In this conformation, Nrf2 is ubiquitinated, released from Keap1 and degraded by the proteasome and free Keap1 is regenerated allowing the cycle to continue. (B) Electrophilic inducers react with cysteine sensors in Keap1 and block the cycle by uncoupling the formation of the ‘closed’ conformation of the Keap1–Nrf2 complex from Nrf2 ubiquitination and degradation. This allows newly translated Nrf2 to accumulate, translocate to the nucleus and activate cytoprotective gene expression.
Exposure to the electrophilic isothiocyanate sulforaphane [1-isothiocyanato-(4R)-(methylsulfinyl)butane] [13] or its derivative sulfoxythiocarbamate alkyne (STCA) [14], two inducers of the Keap1–Nrf2 pathway, which target different cysteine sensors of Keap1, leads to a reduction in the fluorescence lifetime of the donor. Furthermore, these inducers alter the balance between the ‘open’ and ‘closed’ conformations of the Keap1–Nrf2 complex, favouring the ‘closed’ conformation [10,15]. Together with previous data showing that inducers cause conformational changes in Keap1 without dissociation of Nrf2 [16–18], these findings suggest that the inducer-mediated cysteine modifications of Keap1 may alter the positioning of Nrf2 relative to the E2-ubiqitination machinery such that Nrf2 can no longer be ubiquitinated and released from Keap1. Thus, the free Keap1 dimer is not regenerated, the cycle is blocked and the newly translated Nrf2 accumulates and translocates to the nucleus, where it activates transcription of cytoprotective target genes (Figure 2B).
The role of the Keap1–Nrf2 pathway in redox metabolism
The Keap1–Nrf2 pathway is recognized as a master regulator of cellular redox metabolism. Nrf2 directly regulates the gene expression of the catalytic and the regulatory subunits of γ-glutamyl cysteine ligase (GCL), the enzyme catalysing the rate-limiting step in the biosynthesis of GSH [19]. Additionally, the xCT subunit of system xc− that imports cystine into cells, which upon its conversion to cysteine serves as a precursor for the biosynthesis of GSH, is a transcriptional target of Nrf2 [20]. Glutathione peroxidase 2 (GPX2) and glutathione reductase 1 (GSR1), which reduces GSSG, thus maintaining the levels of GSH, are also transcriptionally regulated by Nrf2 [21]. Cells deficient in Nrf2 have lower levels of GSH, whereas Nrf2 activation leads to GSH up-regulation [22]. As GSH is the principal small molecule antioxidant in the cell, the levels of reactive oxygen species (ROS) are increased under conditions of Nrf2 deficiency [23]. Conversely, the levels of ROS are lower when Nrf2 is constitutively activated by genetic or pharmacologic means, particularly upon challenge with agents causing oxidative stress [22,24,25].
In addition to its role in GSH generation and maintenance, Nrf2 controls the expression of the proteins responsible for the reduction in oxidized protein thiols: thioredoxin (TXN) [26], TXN reductase 1 (TXNR1) [27,28] and sulfiredoxin (SRX) [29]. Furthermore, Nrf2 provides the reducing equivalents needed for the maintenance of glutathione and TXN in their reduced states. NADPH is an obligatory cofactor for both GSR and TXNR and Nrf2 is a major contributor to the cellular NADPH levels by regulating the gene expression of all four principal enzymes which generate NADPH: malic enzyme 1 (ME-1), isocitrate dehydrogenase 1 (IDH-1), glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (PGD) [30–33].
Unexpectedly, primary cultures of neuronal cells and brain tissue slices isolated from Keap1-knockdown (Keap1-KD) mice have higher rates of ROS production compared with their WT counterparts, although the magnitude of the difference between these genotypes is not as large as that between Nrf2-knockout (Nrf2-KO) and WT. Analysis of the mRNA expression levels of the two NADPH oxidase (NOX) isoforms implicated in brain pathology showed that NOX2 is dramatically up-regulated under conditions of Nrf2 deficiency (Nrf2-KO), whereas NOX4 is up-regulated when Nrf2 is constitutively activated (Keap1-KD) to a degree which parallels the increases in ROS production [34].
The emerging role of the Keap1–Nrf2 pathway in bioenergetics
Mitochondrial function is a central determinant of cellular redox homoeostasis. Given its role as a master regulator of redox metabolism, it is perhaps not surprising that Nrf2 may affect the activity of this organelle. Indeed, several reports have linked Nrf2 to mitochondrial function. Thus, Nrf2 has been shown to enhance gene expression of transcription factors nuclear respiratory factor 1 and peroxisome proliferator-activated receptor γ coactivator-1α, which in turn promote mitochondrial biogenesis [35,36]. Interestingly, in addition to the cytoplasmic pool, cells also contain a pool of Keap1 that is tethered to the outer mitochondrial membrane forming a ternary complex with Nrf2 and phosphoglycerate mutase 5 (PGAM5), a member of the phosphoglycerate mutase family [37]. Although at a slower rate in comparison with Nrf2, in cancer cells Keap1 is able to mediate the ubiquitination and proteasomal degradation of PGAM5, which in turn is bound to B-cell lymphoma-extra large (Bcl-xl), thereby triggering apoptosis [38,39]. Upon Nrf2 KD in human colon cancer cells, oxygen consumption and ATP production are decreased [40], indicating compromised mitochondrial function.
Considering the breadth of cytoprotection provided by the Keap1–Nrf2 pathway, we recently set out to explore the role of Nrf2 in mitochondrial physiology, focusing initially on one of its most important hallmarks, the mitochondrial membrane potential (ΔΨm). Live cell imaging experiments revealed that in mouse embryonic fibroblasts (MEF) and neurons growing in primary midbrain cultures isolated from WT, Nrf2-KO, Keap1-KO or Keap1-KD mice [41–44], disruption of Nrf2 results in a significant loss of the basal ΔΨm [23]. Conversely, constitutive Nrf2 activation (by KO or KD of Keap1) increases ΔΨm. As the mitochondrial membrane potential is a unique indicator of mitochondrial function, this finding indicates that the Keap1–Nrf2 pathway may be involved in the control of respiration, oxidative phosphorylation and/or mitochondrial redox homoeostasis, all of which depend on ΔΨm.
This Nrf2-mediated metabolic regulation has an impact on mitochondrial ROS production. Thus, Nrf2 deficiency causes a decrease in ∆Ψm due to inhibition of mitochondrial respiration that leads to increased ROS production. Activation of respiration with substrates of respiration restores ∆Ψm and decreases mitochondrial ROS. In contrast, constitutive Nrf2 activation (by KO or KD of Keap1) increases ΔΨm that in turn also increases ROS production in mitochondria. Importantly, application of substrates for the electron transport chain to these cells induces further increase in mitochondrial ROS [34].
The use of inhibitors of oxidative phosphorylation or glycolysis has revealed that Nrf2 affects not only the total levels of ATP [23,40], but also the way by which cells synthesize ATP [23]. Thus, application of oligomycin, an inhibitor of the F1F0-ATP synthase, to WT neurons causes a slight hyperpolarization of the mitochondrial membrane as proton entry through the F1F0-ATP synthase is inhibited. As expected, this is accompanied by a decrease in the ATP levels. In agreement with the predominant use of oxidative phosphorylation for ATP production in neurons, inhibition of glycolysis by application of iodoacetic acid after oligomycin has no further effect on the ATP levels in WT cells. In sharp contrast, oligomycin causes a marked mitochondrial depolarization in Nrf2-KO neurons, accompanied by an increase in the ATP levels, which in turn are decreased by iodoacetic acid. Together, these findings indicate that, under conditions of Nrf2 deficiency, ATP is produced primarily in glycolysis, not oxidative phosphorylation and the ΔΨm is largely maintained by the hydrolysis of ATP by the F1F0-ATPase, rather than by respiration. Such ‘reversal’ of function of the F1F0-ATP synthase towards ATPase activity is observed in the event of mitochondrial damage or inhibition of respiration, allowing cells to maintain ΔΨm by ATP hydrolysis, thus restoring the proton gradient across the membrane. Although the structural integrity of Nrf2-deficient brain mitochondria has not been examined, it is noteworthy that hepatocytes from livers of Nrf2-KO mice that had been fed high-fat diet for 24 weeks contain swollen mitochondria with reduced cristae and disrupted membranes [45].
One reason for the observed impaired respiration in the absence of Nrf2 could be less efficient production of NADH in the tricarboxylic acid (TCA) cycle. Indeed, in Nrf2-KO cells, the mitochondrial pool of NADH is almost 2-fold smaller compared with WT cells and the rate of regeneration of NADH in response to inhibition of respiration is slower [23]. Furthermore, application of substrates for the TCA cycle (pyruvate and malate) has much smaller effects on the ΔΨm and respiration in Nrf2-KO than in WT cells. This implies that, under conditions of Nrf2 deficiency, the activity of complex I in the respiratory chain may be impaired due to limited production/availability of its substrate, NADH.
In addition to complex I, Nrf2 deficiency may also affect complex II-dependent respiration by limiting the generation of FADH2. Similar to NADH, the rate of generation of FADH2 after inhibition of respiration is slower in cultured Nrf2-KO neurons and heart tissue slices compared with their WT counterparts [23,46]. Strikingly, application of methyl succinate, a substrate for complex II, induces a slight depolarization in Nrf2-KO neurons, whereas it causes the expected hyperpolarization in WT cells [23]. Constitutive activation of Nrf2 (by Keap1-KD) leads to the expected increase in the Δψm in response to pyruvate, malate and methyl succinate. Unexpectedly however, the fast increase in the ΔΨm upon addition of these TCA cycle substrates are followed by rapid decreases, suggesting an unusually fast consumption of these substrates. In agreement, the levels of malate, pyruvate and succinate after a 1-h pulse of [U-13C6]-glucose are 50%–70% lower in Keap1-KO compared with WT MEF [33]. Thus, one way by which Nrf2 influences cellular bioenergetics is by controlling substrate availability for mitochondrial respiration.
Curiously, incubation of MEF cells in medium without glucose accentuates the effect of Nrf2 deficiency on the ΔΨm, which under these conditions is nearly ~50% lower than that of the WT control [23]. The glucose dependence of Δψm suggests that Nrf2 is affecting mitochondrial fatty acid oxidation (FAO) as, under conditions of glucose deprivation, FAO becomes a major provider of substrates for respiration and oxidative phosphorylation. If Nrf2 affects FAO, then Nrf2 deletion may lead to mitochondrial dysfunction, decreased ATP synthesis and increased production of ROS. We have therefore evaluated the efficiency of FAO using the long-chain (C16:0) saturated fatty acid, palmitic acid, as well as the short-chain (C6:0) hexanoic acid in MEF of the three (WT, Nrf2-KO and Keap1-KO) genotypes [46]. Application of palmitic or hexanoic acid produces a significantly higher rate of respiration in Keap1-KO MEF than in their WT counterparts, whereas in Nrf2-KO MEF, the rate of respiration is slower compared with WT cells. Experiments using isolated mitochondria from heart and liver of WT, Nrf2-KO and Keap1-KD mice give similar results. These differences among the genotypes show that the efficiency of FAO is influenced by the status of Nrf2, whereby FAO is impaired in the absence of the transcription factor and accelerated under conditions of constitutive Nrf2 activation.
The α-β dehydrogenation of the acyl-CoA fatty acid ester represents the first step in mitochondrial FAO. During deprotonation of the α-carbon, the pro-R hydrogen of the β-carbon leaves as a hydride which reduces the FAD cofactor. The resulting FADH2 then transfers electrons to ubiquinone in the respiratory chain, ultimately contributing to ATP synthesis. As expected, stimulation of FAO by palmitoylcarnitine causes an increase in the ATP levels in WT MEF and the ATP increase is faster in Keap1-KO cells [46]. In sharp contrast, there is no change in the ATP levels in Nrf2-KO MEF upon application of palmitoylcarnitine, confirming that, in the absence of Nrf2, FAO is suppressed and strongly suggesting that the lower ATP levels under conditions of Nrf2 deficiency [23,40] are in part due to suppression of FAO. Additionally, a recent report has shown that KD of Nrf2 in human 293T cells reduces the expression of CPT1 and CPT2 [47], two isoforms of carnitine palmitoyltransferase (CPT). CPT is the enzyme responsible for the rate-limiting step in mitochondrial FAO by catalysing the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to L-carnitine, thereby allowing the transfer of acyl-carnitine from the cytoplasm into the mitochondrial intermembrane space. Thus, another way by which Nrf2 influences cellular bioenergetics is by controlling the efficiency of mitochondrial FAO.
Role of pharmacological activation of Nrf2 in neuronal protection in PINK1 deficiency
Oxidative stress and mitochondrial dysfunction have been implicated in the pathology of Parkinson's disease. Mutations in the mitochondrial serine/threonine-protein kinase PTEN-induced kinase 1 (PINK1) are associated with hereditary early-onset Parkinson's disease [48]. Recently, we found that PINK1 deficiency is associated with inhibition of mitochondrial respiration due to lack of mitochondrial substrates that lead to decrease in ∆Ψm (Figure 3A) [49,50]. Provision of PINK1-deficient cells with mitochondrial substrates restores ∆Ψm and makes these cells less vulnerable to dopamine-induced neurodegeneration [51].
Figure 3. Inducers of the Keap1-Nrf2 pathway restore the mitochondrial membrane potential in PINK1-deficient primary neurons and astrocytes and protects against dopamine-induced cell death.

(A) Primary midbrain neurons and astrocytes isolated from WT and PINK1-KO mice were treated with sulforaphane (50 nM, 24 h) or RTA-408 (20 nM, 24 h) and loaded with 25 nM tetramethylrhodamine methyl ester (TMRM) for 40 min for determination of the mitochondrial membrane potential. (B) Effect of pre-treatment with inducers for 24 h before and during the time (a further 24 h) of exposure of co-cultures of neurons and astrocytes to 50 μM dopamine. Cell death was measured by counting the dead cells (Propidium Iodide, red fluorescence) and live cells (Hoechst 33342, blue fluorescence). Blank indicates cells treated with solvent (0.1% DMSO). *P<0.01; **P<0.001.
The similarity between the effects of PINK1 and Nrf2 deficiency on mitochondrial bioenergetics prompted us to test the hypothesis that Nrf2 inducers may lead to a recovery of mitochondrial metabolism under conditions of PINK1 deficiency. Indeed, incubation of primary co-cultures of midbrain neurons and astrocytes isolated from PINK1-KO mice with the Nrf2 inducers RTA-408 (20 nM), a synthetic triterpenoid [52] or sulforaphane (50 nM) [13] restored the mitochondrial membrane potential in these cells (∆Ψm increased from 84±3.8% of WT to 103.6±4.9% for RTA-408 and to 98.7±6.7% for sulforaphane; Figure 3A). Furthermore, such pharmacological Nrf2 activation was protective against the toxicity of dopamine. Thus, incubation with dopamine (50 μM) for 24 h resulted in an increase in cell death in both WT and PINK1-KO cells, although the level of cell death was greater in PINK1-KO cells (31.1±4.2%, n=7, in WT compared with 59.8±5.2%, n=12, in PINK1-KO cells; P<0.001; Figure 3B). Both RTA-408 (20 nM) and sulforaphane (50 nM) were able to reduce the dopamine-induced cell death in WT and PINK1-KO co-cultures of neurons and astrocytes (Figure 3B). Thus, inducers of Nrf2 restore the mitochondrial membrane potential in PINK1-KO cells and protect against dopamine-induced cell death. These results suggest that pharmacological inducers of Nrf2, by improving mitochondrial function, may be beneficial in PINK1-associated Parkinson's disease as has been reported for the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) animal model of Parkinson's disease [53–56].
Conclusions
The Keap1–Nrf2 pathway is known to orchestrate an elaborate genetic program that enhances the intrinsic cytoprotective functions, including drug metabolizing, antioxidant and anti-inflammatory networks. Dysregulation of Nrf2 activity is causally associated with susceptibility to chronic disease and Nrf2 is now considered a drug target for various pathologies in which oxidative stress and inflammation underlie disease pathogenesis. The emerging role of the Keap1–Nrf2 pathway in cellular bioenergetics adds another layer to the broad cytoprotection which it provides. The role of this pathway in the maintenance of mitochondrial homoeostasis is consistent with the beneficial effects of Nrf2 activators in numerous models of human disease. Particularly exciting are the recent reports showing mitochondrial dysfunction, higher oxidative stress and lower gene expression of Nrf2 in children with autism spectrum disorder [57] and improvements of a number of clinical outcomes in such children upon intervention with the Nrf2 inducer sulforaphane [58]. In addition, metabolic changes indicating enhanced integration of FAO with the activity of the TCA cycle have been observed in human subjects after consumption of diets rich in glucoraphanin, the precursor of sulforaphane [59]. Together, these finding reinforce the notion that the Keap1–Nrf2 pathway is an attractive target for disease prevention and treatment. For the successful realization of this idea, it is essential to understand the details of the regulation of Nrf2 and of the precise mechanisms by which it affects mitochondrial function.
Acknowledgments
We thank all members of our laboratories for their participation in the work summarized in this paper and Reata Pharmaceuticals for providing RTA-408.
Abbreviations
- CPT
carnitine palmitoyltransferase
- E-FRET
FRET efficiency
- FAO
fatty acid oxidation
- GCL
glutamyl cysteine ligase
- GSR
glutathione reductase
- KD
knockdown
- KO
knockout
- Keap1
Kelch-like ECH associated protein 1
- MEF
mouse embryonic fibroblasts
- Nrf2
NF-E2 p45-related factor 2
- PINK1
PTEN-induced kinase 1
- ROS
reactive oxygen species
- TCA
tricarboxylic acid
- TXN
thioredoxin
- TXNR
thioredoxin reductase
- WT
wild-type
Footnotes
The Keap1/Nrf2 Pathway in Health and Disease: Held at Robinson College, Cambridge, UK, 6–8 Jan 2015.
Funding
This work was supported by the Wellcome Trust/MRC Parkinson's Consortium; the MRC doctoral training programme at the University of Dundee [reference number G0800118]; the Cancer Research UK [grant number C20953/A10270]; and the Biotechnology and Biological Sciences Research Council [grant numbers BB/J007498/1 and BB/L01923X/1].
References
- 1.Itoh K., Wakabayashi N., Katoh Y., Ishii T., Igarashi K., Engel J.D., Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cullinan S.B., Gordan J.D., Jin J., Harper J.W., Diehl J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004;24:8477–8486. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobayashi A., Kang M.I., Okawa H., Ohtsuji M., Zenke Y., Chiba T., Igarashi K., Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang D.D., Lo S.C., Cross J.V., Templeton D.J., Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dinkova-Kostova A.T., Holtzclaw W.D., Cole R.N., Itoh K., Wakabayashi N., Katoh Y., Yamamoto M., Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMahon M., Lamont D.J., Beattie K.A., Hayes J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18838–18843. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kensler T.W., Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31:90–99. doi: 10.1093/carcin/bgp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki T., Motohashi H., Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013;34:340–346. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Hayes J.D., Dinkova-Kostova A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Baird L., Lleres D., Swift S., Dinkova-Kostova A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. U.S.A. 2013;110:15259–15264. doi: 10.1073/pnas.1305687110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMahon M., Thomas N., Itoh K., Yamamoto M., Hayes J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006;281:24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 12.Tong K.I., Kobayashi A., Katsuoka F., Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol. Chem. 2006;387:1311–1320. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y., Talalay P., Cho C.G., Posner G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc. Natl. Acad. Sci. U.S.A. 1992;89:2399–2403. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahn Y.H., Hwang Y., Liu H., Wang X.J., Zhang Y., Stephenson K.K., Boronina T.N., Cole R.N., Dinkova-Kostova A.T., Talalay P., Cole P.A. Electrophilic tuning of the chemoprotective natural product sulforaphane. Proc. Natl. Acad. Sci. U.S.A. 2010;107:9590–9595. doi: 10.1073/pnas.1004104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baird L., Swift S., Lleres D., Dinkova-Kostova A.T. Monitoring Keap1-Nrf2 interactions in single live cells. Biotechnol. Adv. 2014;32:1133–1144. doi: 10.1016/j.biotechadv.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinkova-Kostova A.T., Holtzclaw W.D., Wakabayashi N. Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry. 2005;44:6889–6899. doi: 10.1021/bi047434h. [DOI] [PubMed] [Google Scholar]
- 17.Eggler A.L., Liu G., Pezzuto J.M., van Breemen R.B., Mesecar A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. U.S.A. 2005;102:10070–10075. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y., Paonessa J.D., Zhang Y. Mechanism of chemical activation of Nrf2. PLoS One. 2012;7:e35122. doi: 10.1371/journal.pone.0035122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wild A.C., Moinova H.R., Mulcahy R.T. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 20.Sasaki H., Sato H., Kuriyama-Matsumura K., Sato K., Maebara K., Wang H., Tamba M., Itoh K., Yamamoto M., Bannai S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002;277:44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- 21.Singh A., Rangasamy T., Thimmulappa R.K., Lee H., Osburn W.O., Brigelius-Flohe R., Kensler T.W., Yamamoto M., Biswal S. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am. J. Respir Cell Mol. Biol. 2006;35:639–650. doi: 10.1165/rcmb.2005-0325OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benedict A.L., Knatko E.V., Dinkova-Kostova A.T. The indirect antioxidant sulforaphane protects against thiopurine-mediated photooxidative stress. Carcinogenesis. 2012;33:2457–2466. doi: 10.1093/carcin/bgs293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmstrom K.M., Baird L., Zhang Y., Hargreaves I., Chalasani A., Land J.M., Stanyer L., Yamamoto M., Dinkova-Kostova A.T., Abramov A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open. 2013;2:761–770. doi: 10.1242/bio.20134853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao X., Dinkova-Kostova A.T., Talalay P. Powerful and prolonged protection of human retinal pigment epithelial cells, keratinocytes, and mouse leukemia cells against oxidative damage: the indirect antioxidant effects of sulforaphane. Proc. Natl. Acad. Sci. U.S.A. 2001;98:15221–15226. doi: 10.1073/pnas.261572998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalra S., Knatko E.V., Zhang Y., Honda T., Yamamoto M., Dinkova-Kostova A.T. Highly potent activation of Nrf2 by topical tricyclic bis(cyano enone): implications for protection against UV radiation during thiopurine therapy. Cancer Prev. Res. 2012;5:973–981. doi: 10.1158/1940-6207.CAPR-12-0041. [DOI] [PubMed] [Google Scholar]
- 26.Hawkes H.J., Karlenius T.C., Tonissen K.F. Regulation of the human thioredoxin gene promoter and its key substrates: a study of functional and putative regulatory elements. Biochim. Biophys. Acta. 2014;1840:303–314. doi: 10.1016/j.bbagen.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 27.Sakurai A., Nishimoto M., Himeno S., Imura N., Tsujimoto M., Kunimoto M., Hara S. Transcriptional regulation of thioredoxin reductase 1 expression by cadmium in vascular endothelial cells: role of NF-E2-related factor-2. J. Cell Physiol. 2005;203:529–537. doi: 10.1002/jcp.20246. [DOI] [PubMed] [Google Scholar]
- 28.Wakabayashi N., Dinkova-Kostova A.T., Holtzclaw W.D., Kang M.I., Kobayashi A., Yamamoto M., Kensler T.W., Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. U.S.A. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abbas K., Breton J., Planson A.G., Bouton C., Bignon J., Seguin C., Riquier S., Toledano M.B., Drapier J.C. Nitric oxide activates an Nrf2/sulfiredoxin antioxidant pathway in macrophages. Free Radic. Biol. Med. 2011;51:107–114. doi: 10.1016/j.freeradbiomed.2011.03.039. [DOI] [PubMed] [Google Scholar]
- 30.Thimmulappa R.K., Mai K.H., Srisuma S., Kensler T.W., Yamamoto M., Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 31.Lee J.M., Calkins M.J., Chan K., Kan Y.W., Johnson J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 32.Wu K.C., Cui J.Y., Klaassen C.D. Beneficial role of nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011;123:590–600. doi: 10.1093/toxsci/kfr183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 34.Kovac S., Angelova P.R., Holmstrom K.M., Zhang Y., Dinkova-Kostova A.T., Abramov A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta. 2015;1850:794–801. doi: 10.1016/j.bbagen.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piantadosi C.A., Carraway M.S., Babiker A., Suliman H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 2008;103:1232–1240. doi: 10.1161/01.RES.0000338597.71702.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacGarvey N.C., Suliman H.B., Bartz R.R., Fu P., Withers C.M., Welty-Wolf K.E., Piantadosi C.A. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am. J. Respir. Crit. Care Med. 2012;185:851–861. doi: 10.1164/rccm.201106-1152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lo S.C., Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp. Cell Res. 2008;314:1789–1803. doi: 10.1016/j.yexcr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo S.C., Hannink M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J. Biol. Chem. 2006;281:37893–37903. doi: 10.1074/jbc.M606539200. [DOI] [PubMed] [Google Scholar]
- 39.Xu Y., Fang F., Miriyala S., Crooks P.A., Oberley T.D., Chaiswing L., Noel T., Holley A.K., Zhao Y., Kiningham K.K., et al. KEAP1 is a redox sensitive target that arbitrates the opposing radiosensitive effects of parthenolide in normal and cancer cells. Cancer Res. 2013;73:4406–4417. doi: 10.1158/0008-5472.CAN-12-4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim T.H., Hur E.G., Kang S.J., Kim J.A., Thapa D., Lee Y.M., Ku S.K., Jung Y., Kwak M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res. 2011;71:2260–2275. doi: 10.1158/0008-5472.CAN-10-3007. [DOI] [PubMed] [Google Scholar]
- 41.Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 42.Wakabayashi N., Itoh K., Wakabayashi J., Motohashi H., Noda S., Takahashi S., Imakado S., Kotsuji T., Otsuka F., Roop D.R., et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 43.Taguchi K., Maher J.M., Suzuki T., Kawatani Y., Motohashi H., Yamamoto M. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol. Cell. Biol. 2010;30:3016–3026. doi: 10.1128/MCB.01591-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higgins L.G., Hayes J.D. The cap’n’collar transcription factor Nrf2 mediates both intrinsic resistance to environmental stressors and an adaptive response elicited by chemopreventive agents that determines susceptibility to electrophilic xenobiotics. Chem. Biol. Interact. 2011;192:37–45. doi: 10.1016/j.cbi.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 45.Meakin P.J., Chowdhry S., Sharma R.S., Ashford F.B., Walsh S.V., McCrimmon R.J., Dinkova-Kostova A.T., Dillon J.F., Hayes J.D., Ashford M.L. Susceptibility of Nrf2-null mice to Steatohepatitis and Cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014;34:3305–3320. doi: 10.1128/MCB.00677-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ludtmann M.H., Angelova P.R., Zhang Y., Abramov A.Y., Dinkova-Kostova A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014;457:415–424. doi: 10.1042/BJ20130863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pang S., Lynn D.A., Lo J.Y., Paek J., Curran S.P. SKN-1 and Nrf2 couples proline catabolism with lipid metabolism during nutrient deprivation. Nat. Commun. 2014;5:5048. doi: 10.1038/ncomms6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Valente E.M., Abou-Sleiman P.M., Caputo V., Muqit M.M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A.R., Healy D.G., et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 49.Gandhi S., Wood-Kaczmar A., Yao Z., Plun-Favreau H., Deas E., Klupsch K., Downward J., Latchman D.S., Tabrizi S.J., Wood N.W., et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abramov A.Y., Gegg M., Grunewald A., Wood N.W., Klein C., Schapira A.H. Bioenergetic consequences of PINK1 mutations in Parkinson disease. PLoS One. 2011;6:e25622. doi: 10.1371/journal.pone.0025622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gandhi S., Vaarmann A., Yao Z., Duchen M.R., Wood N.W., Abramov A.Y. Dopamine induced neurodegeneration in a PINK1 model of Parkinson's disease. PLoS One. 2012;7:e37564. doi: 10.1371/journal.pone.0037564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reisman S.A., Lee C.Y., Meyer C.J., Proksch J.W., Ward K.W. Topical application of the synthetic triterpenoid RTA 408 activates Nrf2 and induces cytoprotective genes in rat skin. Arch. Dermatol. Res. 2014;306:447–454. doi: 10.1007/s00403-013-1433-7. [DOI] [PubMed] [Google Scholar]
- 53.Burton N.C., Kensler T.W., Guilarte T.R. In vivo modulation of the Parkinsonian phenotype by Nrf2. Neurotoxicology. 2006;27:1094–1100. doi: 10.1016/j.neuro.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 54.Chen P.C., Vargas M.R., Pani A.K., Smeyne R.J., Johnson D.A., Kan Y.W., Johnson J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Innamorato N.G., Jazwa A., Rojo A.I., Garcia C., Fernandez-Ruiz J., Grochot-Przeczek A., Stachurska A., Jozkowicz A., Dulak J., Cuadrado A. Different susceptibility to the Parkinson's toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS One. 2010;5:e11838. doi: 10.1371/journal.pone.0011838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaidery N.A., Banerjee R., Yang L., Smirnova N.A., Hushpulian D.M., Liby K.T., Williams C.R., Yamamoto M., Kensler T.W., Ratan R.R., et al. Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson's disease. Antioxid. Redox Signal. 2013;18:139–157. doi: 10.1089/ars.2011.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Napoli E., Wong S., Hertz-Picciotto I., Giulivi C. Deficits in bioenergetics and impaired immune response in granulocytes from children with autism. Pediatrics. 2014;133:e1405–1410. doi: 10.1542/peds.2013-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh K., Connors S.L., Macklin E.A., Smith K.D., Fahey J.W., Talalay P., Zimmerman A.W. Sulforaphane treatment of autism spectrum disorder (ASD) Proc. Natl. Acad. Sci. U.S.A. 2014;111:15550–15555. doi: 10.1073/pnas.1416940111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Armah C.N., Traka M.H., Dainty J.R., Defernez M., Janssens A., Leung W., Doleman J.F., Potter J.F., Mithen R.F. A diet rich in high-glucoraphanin broccoli interacts with genotype to reduce discordance in plasma metabolite profiles by modulating mitochondrial function. Am. J. Clin. Nutr. 2013;98:712–722. doi: 10.3945/ajcn.113.065235. [DOI] [PMC free article] [PubMed] [Google Scholar]