

The identification or prediction of functional regulatory elements across the genome is of fundamental importance in biology. Recent studies have demonstrated that the genome is pervasively transcribed, giving rise to thousands of noncoding RNAs (ncRNAs), many of which originate from regulatory elements, including transcription factor binding sites (TFBSs; i.e., enhancers).1,2 The relationship between the expression of these ncRNAs and the function of the regulatory elements is poorly understood. Nonetheless, the expression of overlapping ncRNAs is a good mark of active enhancers and can be used in enhancer prediction.3 In the current issue of Cell Cycle, Vucicevic et al. describe the use of a computational enhancer prediction tool called PreSTIGE to integrate information about ncRNA expression at enhancers with information about tissue-specific target gene expression, allowing prediction of functional enhancers.4 This method represents an improvement over enhancer predictions based solely on histone modifications.
Enhancer-originating ncRNAs include long non-coding RNAs (lncRNAs) and enhancer RNAs (eRNAs).1–3 LncRNAs are similar in many respects to protein coding mRNAs (mRNAs); they are 5’ capped, spliced, and 3’ polyadenylated. Unlike mRNAs, however, lncRNAs exhibit poor evolutionary conservation, limited coding potential, and very highly cell type-specific expression patterns.2 eRNAs are short, usually bi-directional transcripts that originate at or near TFBSs.1,3 The functional distinction between enhancer-originating lncRNAs (called activating ncRNAs or ncRNA-a) and eRNAs, if any, is unclear. Furthermore, their enhancer-regulating functions as RNA molecules, as opposed to byproducts of regulatory transcription at TFBSs, is still debated in the literature.2,3 Nonetheless, as shown by Vucicevic et al. for ncRNA-a,4 as well as by others for eRNAs,3 the expression of these ncRNAs can be used as a mark to identify active enhancers.
PreSTIGE predicts enhancers by first identifying mRNA genes with elevated tissue-specific expression as likely targets of tissue-specific enhancers. It then identifies from ChIP-seq data histone H3 lysine 4 monomethyl (H3K4me1) domains in the vicinity, which are used as a mark for enhancers. In this way, the predicted enhancers are linked with the tissue-specific expression of nearby putative target genes.5 In their recent study, Vucicevic et al. take this approach one step further by examining lncRNA transcription overlapping the H3K4me1-predicted enhancers (Fig. 1).4 Specifically, they investigated the overlap between ∼9,500 ENCODE-annotated lncRNAs and ∼132,000 PreSTIGE-predicted cell-type specific enhancers. They observed that ∼2,700 of the lncRNAs (∼28% of the total analyzed) overlap a predicted cell type-specific enhancer in any of the 11 cell lines used as input into the prediction algorithm. These results suggest that (1) cell type-specific enhancers associated with cell type-specific gene expression represent a small fraction of the overall enhancer repertoire in the cell and (2) enhancer lncRNA transcription is functionally linked to cell type-specific target gene expression. Although a positive functional link is assumed, previous studies suggest this is not always the case (e.g., the well-characterized lncRNA HOTAIR represses the expression of neighboring genes6), an issue that could limit the use of this approach.
In addition to improving enhancer prediction, the approach from Vucicevic et al. also reveals some interesting features of enhancers. For example, enhancers overlapping an annotated lncRNA have relatively higher H3K4me3/H3K4me1 ratios compared to enhancers that do not overlap an annotated lncRNA. The enrichment of H3K4me3, a mark associated with actively transcribed promoters, likely reflects the transcription activity of overlapping expressed lncRNA genes. Furthermore, Vucicevic et al.4 observed a strong correlation between the expression levels of lncRNAs overlapping cell type-specific enhancers and the protein-coding genes that are targeted by those enhancers, hinting at a functional link between the two. The target protein-coding genes are enriched for ontological terms relevant to the biology of the corresponding cell types, suggesting that the approach identifies functionally relevant enhancers. In spite of this strong correlation, the functional and mechanistic relationships between enhancers, their overlapping lncRNAs, and the target protein-coding genes remain to be determined.
Although the calling of enhancers that are likely functional is improved by taking the expression of enhancer-originating lncRNAs and nearby target genes into account, the total number of enhancers defined in this way is only a fraction of the enhancers called by ChIP-seq (e.g., using H3K4me1).4 In addition to the cell type specificity noted above, this may also be due, in part, to a failure to detect enhancers that are located distal to the target genes. Determining the effectiveness of the PreSTIGE-based approach will require a direct comparison with other approaches, including those based on other types of genomic data (e.g., p300 and Mediator ChIP-seq; GRO-seq, PRO-seq, and GRO-cap), function-based screening (e.g., STAR-seq), and alternate computational algorithms (e.g., dREG) (Fig. 1). Considering the pros and cons of all of the available enhancer prediction approaches described in the literature, the best strategy would seem to be a comprehensive approach that is designed specifically for the available data and the particular biological question. The current approaches will undoubtedly be improved by the integration of additional genomic information, such as enhancer-promoter looping by chromosome conformation capture.7 And, of course, all predictions should ultimately be followed by in depth experimental validation, including enhancer deletion.
Figure 1.
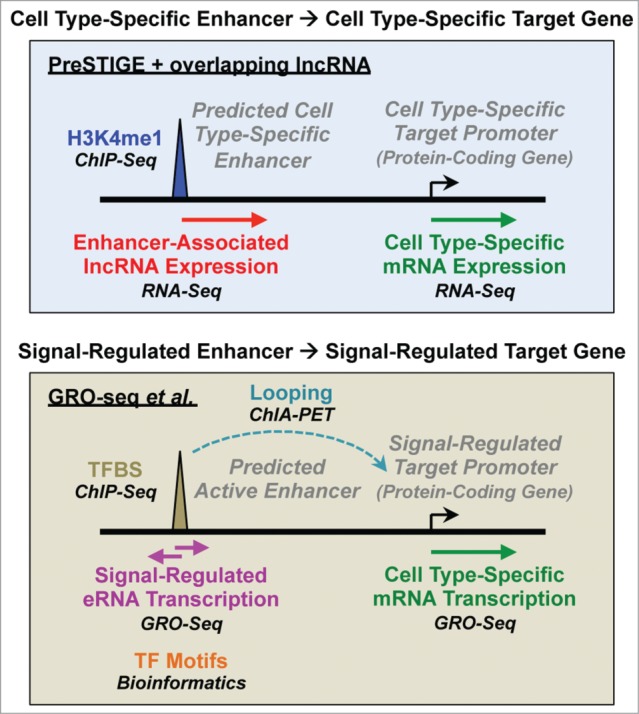
Different approaches for the integration of genomic data to predict functional enhancers. Various types of genomic data can be integrated using computational pipelines, such as PreSTIGE,5 to predict functional enhancers. Vucicevic et al.4 used PreSTIGE in conjunction with enhancer-associated lncRNA expression to identify cell type-specific enhancers (top), while Hah et al.3 used a pipeline that integrates enhancer and target gene transcription with gene looping to predict functional estrogen-regulated enhancers (bottom).
References
- 1.Hah N, Kraus WL. Mol Cell Endocrinol 2014; 382:652–64; PMID:23810978; http://dx.doi.org/ 10.1016/j.mce.2013.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonasio R, Shiekhattar R. Annu Rev Genet 2014; 48:433–55; PMID:25251851; http://dx.doi.org/ 10.1146/annurev-genet-120213-092323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hah N, et al.. Genome Res 2013; 23:1210–23; PMID:23636943; http://dx.doi.org/ 10.1101/gr.152306.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vucicevic D, et al.. Cell Cycle 2015; 14:253–60; PMID:25607649; http://dx.doi.org/ 10.4161/15384101.2014.977641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corradin O, et al.. Genome Res 2014; 24:1–13; PMID:24196873; http://dx.doi.org/ 10.1101/gr.164079.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta RA, et al.. Nature 2010; 464:1071–6; PMID:20393566; http://dx.doi.org/ 10.1038/nature08975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma W, et al.. Nat Methods 2015; 12:71–8; PMID:25437436; http://dx.doi.org/ 10.1038/nmeth.3205 [DOI] [PMC free article] [PubMed] [Google Scholar]