

Abstract
Sulphonylureas stimulate insulin secretion from pancreatic β-cells primarily by closing ATP-sensitive K+ channels in the β-cell plasma membrane. The mechanism of channel inhibition by these drugs is unusually complex. As direct inhibitors of channel activity, sulphonylureas act only as partial antagonists at therapeutic concentrations. However, they also exert an additional indirect inhibitory effect via modulation of nucleotide-dependent channel gating. In this review, we summarize current knowledge and recent advances in our understanding of the molecular mechanism of action of these drugs.
Keywords: ABCC8, diabetes, KATP channels, Kir6.2, SUR1
Introduction
Sulphonylurea drugs have been used for over 60 years to treat type 2 diabetes [1]. These drugs share a common mechanism of action, which is also the case for the previously developed anti-hyperglycaemic drugs, glinides [2]. Their primary target is the ATP-sensitive potassium (KATP) channel which controls plasma membrane potential in the pancreatic β-cells [3]. Binding of sulphonylureas to KATP channel induces channel closure [4]. This causes membrane depolarization, which activates voltage-dependent Ca2+ channels in the β-cell plasma membrane and the resulting Ca2+ influx triggers Ca2+-dependent insulin granule exocytosis.
In addition to their action on the KATP channel, sulphonylureas can also stimulate insulin secretion independently of changing the membrane potential via interactions with proteins within the exocytotic pathway [5,6].
The B-cell KATP channel and sulphonylureas
The β-cell KATP channel couples glucose metabolism to β-cell electrical activity. Gating (opening and closing) of these channels is primarily determined by intracellular concentrations of adenosine nucleotides (nts). Under Mg2+-free conditions, unchelated ATP (and ADP) inhibits KATP channels through binding to the pore-forming subunit of the channel complex; it is important to note that this inhibitory ligand-binding site does not discriminate between the chelated and unchelated forms of ATP. On the contrary, chelated MgADP (or MgATP) stimulates channel activity through specific interaction with the nt-binding domains of a regulatory subunit of the channel [(sulphonylurea receptor (SUR)], counteracting these nts’ simultaneous inhibitory effect at the channel pore [7].
The KATP channel is a hetero-octameric complex composed of four Kir6.2 subunits and four SUR subunits [8]. Kir6.2 (KCNJ11) is a member of the inwardly rectifying K-channel family; it possesses a potassium-selective pore and an inhibitory site for (Mg)ATP [9]. SURs are members of the C subfamily of the ABC (ATP-binding cassette) superfamily [10]. There are three isoforms of the SUR: ABCC8/SUR1 is expressed in pancreas and neurons, ABCC9/SUR2A in skeletal and cardiac muscle and a splice variant of ABCC9, called SUR2B, in smooth muscle and brain. The major regulatory role of SURs is to mediate stimulation by Mg–adenosine nts via their two nt-binding domains: nt-binding domain 1 (NBD1) and nt-binding domain 2 (NBD2). Nt binding to SURs causes head-to-tail dimerization of the NBDs and formation of two nt-binding sites [NBS1 (nt-binding site 1) and NBS2 (nt-binding site 2)] within the dimer interface. NBS2 possesses greater ATPase activity than NBS1; and its occupancy by MgADP stimulates KATP channel activity [11]. Available data indicate that MgATP promotes channel opening only via its hydrolysis to MgADP [11,12,13]; in this aspect, regulation of KATP channel activity by NBSs of SUR differs from that of other ABC proteins such as CFTR (cystic fibrosis transmembrane conductance regulator) (ABCC7) or P-gp (P-gap, ABCB1) where conformational changes induced by MgATP binding [14–17] result in promotion of channel opening (CFTR) or stimulation of drug transport (P-gp).
KATP channels possess both high-affinity- and low-affinity-binding sites for sulphonylureas and glinides. The precise locations of these binding sites are currently unknown. The high affinity site involves intracellular loops of SUR between transmembrane domains (TMs) 5 and 6 [18] and TMs 15 and 16 [19]. N-terminus of Kir6.2 is also involved in binding of the sulphonylurea glibenclamide and the glinide repaglinide [18,20,21].
Direct inhibitory effect of sulphonylureas
Consistent with the existence of two distinct binding sites, sulphonylurea concentration-inhibition curve for KATP channels measured in excised patches in the absence of nts is biphasic (Figure 1A). The low-affinity block resides on Kir6.2 subunit; the high-affinity site is present only when Kir6.2 is co-expressed with SUR [22]. The low-affinity site is of no clinical relevance as it is occupied only at concentrations much higher than those found in the plasma of patients treated with sulphonylureas [23]. In the absence of intracellular nts, the maximal extent of high-affinity inhibition is only 50%–80%, depending on the type of sulphonylurea [2,4]. Thus, sulphonylureas bound to the high-affinity site act only as partial antagonists so that the channel remains open for a substantial fraction of time when the high-affinity-binding sites are fully occupied.
Figure 1. Direct and indirect inhibitory effects of sulphonylureas.

(A) Gliclazide concentration-inhibition curves for wild-type β-cell KATP currents in excised patch (nt-free solution) and in intact cell (nt present), as indicated. The data is plotted as the current measured in the presence of the drug (I) expressed as a fraction of current in its absence (IC). The fitting parameters were published previously in Proks et al. [4]. The line for data obtained for excised patches is drawn according to equation: 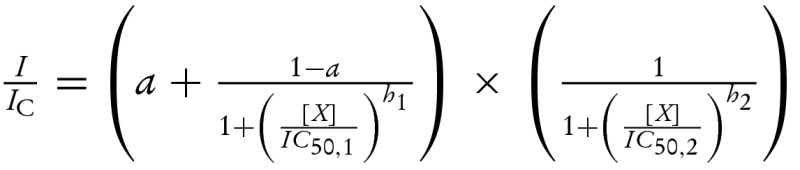 where IC50,1=51 nM and IC50,2=3.1 mM are the half-maximal inhibitory concentrations at the high- and low-affinity sites respectively; h1= 1.0 and h2= 1.0 are the Hill coefficients for the high- and low-affinity sites respectively; and a= 0.4 is the fraction of sulphonylurea-resistant current when all high-affinity sites are occupied. The line for data obtained for intact cell is drawn according to the below equation [4]:
where IC50,1=51 nM and IC50,2=3.1 mM are the half-maximal inhibitory concentrations at the high- and low-affinity sites respectively; h1= 1.0 and h2= 1.0 are the Hill coefficients for the high- and low-affinity sites respectively; and a= 0.4 is the fraction of sulphonylurea-resistant current when all high-affinity sites are occupied. The line for data obtained for intact cell is drawn according to the below equation [4]: 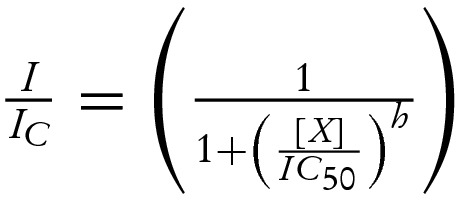 where IC50=108 nM is the half-maximal inhibitory concentration and h=0.81 is the Hill coefficient. (B) Relationship between the fraction of gliclazide-resistant current produced by the direct high-affinity inhibition of the drug (mechanism I in A) and single-channel PO simulated for KATP currents with a simple concerted gating model (Monod–Wyman–Changeux model) [25]. The PO of wild-type KATP channels in excised patches after fast rundown was taken as 0.4 [24]. PO is primary determined by the stability of channel open state; it can be reduced by factors such as channel rundown or increased by channel mutations or via Mg-nt activation of the channel [24,35]. (C and D) Schematic showing effects of nts (C and D) and of nts and sulphonylureas (D) on the wild-type β-cell KATP channel. For clarity, all four subunits of the Kir6.2 tetramer but only one SUR1 subunit (out of the four) are shown. ATP binding (both chelated and unchelated form) to Kir6.2 promotes channel closure whereas MgADP binding to SUR1 promotes channel opening. MgADP antagonizes sulphonylurea binding to SUR1 and vice versa; at therapeutic drug concentrations (at which drug binding to SUR1 is nearly saturated), sulphonylurea effect is dominant. The resulting suppression of MgADP activation ‘unmasks’ the inhibitory effect of ATP on Kir6.2 which enhances that of the sulphonylurea (compare curves obtained for excised patches in nt-free solutions and for intact cells in A).
where IC50=108 nM is the half-maximal inhibitory concentration and h=0.81 is the Hill coefficient. (B) Relationship between the fraction of gliclazide-resistant current produced by the direct high-affinity inhibition of the drug (mechanism I in A) and single-channel PO simulated for KATP currents with a simple concerted gating model (Monod–Wyman–Changeux model) [25]. The PO of wild-type KATP channels in excised patches after fast rundown was taken as 0.4 [24]. PO is primary determined by the stability of channel open state; it can be reduced by factors such as channel rundown or increased by channel mutations or via Mg-nt activation of the channel [24,35]. (C and D) Schematic showing effects of nts (C and D) and of nts and sulphonylureas (D) on the wild-type β-cell KATP channel. For clarity, all four subunits of the Kir6.2 tetramer but only one SUR1 subunit (out of the four) are shown. ATP binding (both chelated and unchelated form) to Kir6.2 promotes channel closure whereas MgADP binding to SUR1 promotes channel opening. MgADP antagonizes sulphonylurea binding to SUR1 and vice versa; at therapeutic drug concentrations (at which drug binding to SUR1 is nearly saturated), sulphonylurea effect is dominant. The resulting suppression of MgADP activation ‘unmasks’ the inhibitory effect of ATP on Kir6.2 which enhances that of the sulphonylurea (compare curves obtained for excised patches in nt-free solutions and for intact cells in A).
In excised patches, KATP channels undergo rundown, spontaneous decline of channel activity, which consists of a fast, dramatic decrease in current during the first minute after patch excision followed by a subsequent slow phase with a mild decrease in current [24]. Figure 1(B) illustrates how rundown may affect the direct inhibitory effect of sulphonylureas; it depicts the dependence of the fraction of drug-resistant current on single channel open probability (PO) for high-affinity gliclazide inhibition; simulated with a simple concerted gating model [25]. The value of intrinsic PO before rundown (or in the intact cell) is unknown; however, it is likely to be much higher than that during the slow phase of rundown during which experiments are usually conducted (PO ∼0.2–0.4) [24]. Thus, it is important to note that the efficacy of direct sulphonylurea inhibition of KATP channels in vivo is likely to be lower than that obtained from data on excised patches.
Indirect inhibitory effect of sulphonylureas
KATP channel activity inside intact pancreatic β-cells is quite low (PO < 0.2) [13] due to the dominant inhibitory effect of large intracellular ATP concentrations. Assuming similar sulphonylurea-binding affinity for nt-bound and nt-free states of the channel, these drugs would produce only partial inhibition of KATP current in intact β-cells (e.g., ∼80% for gliclazide in Figure 1B). Yet, when sulphonylurea drugs are applied to whole-cell KATP currents in intact cells, the high-affinity inhibition is virtually complete (Figure 1A) [2,4].
Multiple studies demonstrated that the maximal extent of high-affinity sulphonylurea inhibition of KATP channels is enhanced in the presence of MgADP in excised patches [2,4,22,26–30] and studies of this effect eventually led to a postulation of a second, indirect action of sulphonylureas on KATP channel gating (Figures 1C and 1D) [2,4,22]. As well as directly inhibiting the channel, sulphonylurea binding to the high-affinity site also suppresses the activatory effect of MgADP (or MgATP); this in turn ‘unmasks’ the inhibitory effect of the nt which is then combined with that of the sulphonylurea (Figures 1A, 1C and 1D).
More recently, the mechanism of sulphonylureas on KATP channel activation by Mg–nts was examined in more detail using KATP channels with a point mutation G334D in the Kir6.2 subunit, Kir6.2–G334D/SUR1 channels (Figure 2) [30]. This mutation severely impairs the inhibitory effect of ATP at Kir6.2 [30–32] which allows studying the properties of Mg–nt activation in isolation from nt inhibition. As shown in Figures 2C and 2D, in the presence of 30 μM gliclazide, concentration-activation curves for both MgADP and MgATP on Kir6.2–G334D/SUR1 channels are dramatically shifted to the right. In addition, the drug also appears to reduce the maximal extent of the activatory effect of the nts. Detailed analysis of data suggest that the drug both reduces nt binding to SUR1 and impairs the efficacy with which nt binding is transduced into pore opening [30].
Figure 2. Suppression of the stimulatory effect of Mg-nts studied with Kir6.2–G334D/SUR1 channels.

(A and B) Schematic showing effects of nts (A and B) and of nts and sulphonylureas (B) on the Kir6.2–G334D/SUR1 channel in which the inhibitory effect of ATP at Kir6.2 is severely impaired. For clarity, all four subunits of the Kir6.2 tetramer but only one SUR1 subunit (out of the four) are shown. MgADP binding to SUR1 promotes channel opening. MgADP antagonizes sulphonylurea binding to SUR1 and vice versa; at therapeutic drug concentrations (at which drug binding to SUR1 is nearly saturated), sulphonylurea effect is dominant. This weakens nt binding and impairs the efficacy with which MgADP binding to SUR1 promotes channel opening (see data depicted in C and D). (C and D) Concentration-activation relationships for MgADP (C) or MgATP (D) for Kir6.2–G334D/SUR1 channels in the absence (open circles) or presence (filled circles) of 30 μM gliclazide. The data are taken from Proks et al. [30]. The lines are the best fit to the mean data of equation: 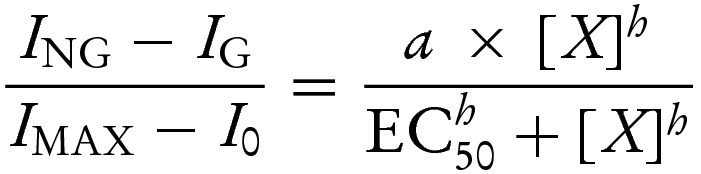 where EC50 is the nt concentration at which activation is half maximal, [X] is the test nt concentration, h is the Hill coefficient and a is the ratio of the maximal stimulatory effect of the nt in the presence and absence of gliclazide. (C) EC50=9 μM, h=1.3; a was fixed at 1 (open circles); EC50=560 μM, h=1.5, a=0.3 (filled circles). (D) EC50=124 μM, h=1.3; a was fixed at 1 (open circles); EC50=8.1 mM, h=1.3; a was set to 0.3 (filled circles).
where EC50 is the nt concentration at which activation is half maximal, [X] is the test nt concentration, h is the Hill coefficient and a is the ratio of the maximal stimulatory effect of the nt in the presence and absence of gliclazide. (C) EC50=9 μM, h=1.3; a was fixed at 1 (open circles); EC50=560 μM, h=1.5, a=0.3 (filled circles). (D) EC50=124 μM, h=1.3; a was fixed at 1 (open circles); EC50=8.1 mM, h=1.3; a was set to 0.3 (filled circles).
Interestingly, a similar effect to gliclazide on MgADP binding and transduction was observed for mutations of either of the Walker A lysines in the catalytic site of the NBS1 or NBS2 of SUR1 [30]. In contrast, these mutations did not appear to impair MgATP binding. This is consistent with a previous finding that mutations of the Walker A lysines reduce, but do not affect Km values of MgATP hydrolysis in SUR1 [33]. In addition to altering the maximal extent of high-affinity sulphonylurea block, Mg–nts also increase the value of IC50 for high-affinity drug inhibition [4,25]. This can be attributed to a reciprocal interaction between the two compounds, displacement of sulphonylurea binding by the nt, as previously demonstrated by Hambrock et al. [34].
Unlike for SUR1-containing channels, Mg–nt activation of KATP channels with the SUR2A regulatory subunit is unaffected by sulphonylureas [25] and the maximal extent of high-affinity sulphonylurea inhibition of SUR2A-containing channels is reduced, rather than increased in the presence of the nt [2,25,35]. This may be a contributing factor to a low occurrence of cardiac side effects of sulphonylureas. Reduced sulphonylurea efficacy in this case may be an indirect effect of Mg–nts on channel gating (Figure 1B).
Implications for treatment of neonatal diabetes
The fact that efficient sulphonylurea inhibition of KATP channels requires the inhibitory effects of nts to be intact is of special significance for treating neonatal diabetes (ND) caused by mutations in the KATP channel [36].
ND mutations are found in both Kir6.2 and SUR1 subunits of the KATP channel [36,37]. Most of these mutations impair channel inhibition by ATP either directly, via reducing ATP binding to Kir6.2, or indirectly, via impairing channel gating [36,37]. The latter mutations stabilize the open state of the channel and thus impair channel ability to close both in the absence and in the presence of nts. Both these defects also affect sulphonylurea efficacy to close the KATP channel; mutations that reduce ATP binding to Kir6.2 impair the indirect inhibitory effect of sulphonylureas whereas mutations that impair ATP inhibition indirectly via affecting channel gating reduce both direct and indirect inhibitory effect of sulphonylureas [25,38,39].
The ability of sulphonylureas to treat patients with ND caused by mutations in KATP channels depends on the severity of functional defect caused by these mutations. This is illustrated in Figure 3 for mutations that impair ATP binding (left, A and C) and channel gating (right, B and D).
Figure 3. Gliclazide inhibition of KATP channels with mutations impairing ATP binding (left) and channel gating (right).

(A–D) Gliclazide concentration–inhibition relations for various mutant channels in the absence (open circles) and presence (filled circles) of various MgATP concentrations. Currents are expressed relative to those in the absence of both MgATP and gliclazide (thus note the different initial current values for different channel mutants in the presence of the nt). The lines are the best fit of the data to the equation: 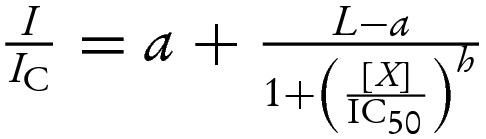 where I is the steady-state KATP current in the presence of the test drug concentration [X], IC is the current in drug free solution obtained by averaging the current before and after application, IC50 is the drug concentration at which the inhibition is half maximal, h is the Hill coefficient, L is a scaling factor reflecting the difference between channel activity in control and MgATP-containing solution (L=1 in the absence of the nt) and a is the fraction of KATP current remaining at gliclazide concentrations that saturate the high-affinity-binding site on SUR1. (A) IC50=49 nM, h=1.2, a=0.48 (open symbols); IC50=190 nM, h=1.2; a=0.25, L=0.96 (filled symbols). (B) IC50=200 nM, h=0.92; a=0.79 (open symbols); IC50=140 nM, h=0.88, a=0.31, L=0.77 (filled symbols). (C) IC50=67 nM, h=1.1; a=0.39 (open symbols); IC50=213 nM, h=1.0, a=0.39, L=2.0 (filled symbols). (D) IC50=930 nM, h=1.5; a=0.95 (open symbols); IC50=1200 nM, h=0.98, a=0.38, L=0.92 (filled symbols). The data were published previously by Proks et al. [25]. (E and F) Comparison of the residual KATP currents in the presence of 100 μM MgATP (E) and 1 mM MgATP (F) in the absence (open bars) and presence of 30 μM gliclazide (filled bars). Currents are expressed relative to those in the absence of both MgATP and gliclazide. The data were published previously by Proks et al. [25].
where I is the steady-state KATP current in the presence of the test drug concentration [X], IC is the current in drug free solution obtained by averaging the current before and after application, IC50 is the drug concentration at which the inhibition is half maximal, h is the Hill coefficient, L is a scaling factor reflecting the difference between channel activity in control and MgATP-containing solution (L=1 in the absence of the nt) and a is the fraction of KATP current remaining at gliclazide concentrations that saturate the high-affinity-binding site on SUR1. (A) IC50=49 nM, h=1.2, a=0.48 (open symbols); IC50=190 nM, h=1.2; a=0.25, L=0.96 (filled symbols). (B) IC50=200 nM, h=0.92; a=0.79 (open symbols); IC50=140 nM, h=0.88, a=0.31, L=0.77 (filled symbols). (C) IC50=67 nM, h=1.1; a=0.39 (open symbols); IC50=213 nM, h=1.0, a=0.39, L=2.0 (filled symbols). (D) IC50=930 nM, h=1.5; a=0.95 (open symbols); IC50=1200 nM, h=0.98, a=0.38, L=0.92 (filled symbols). The data were published previously by Proks et al. [25]. (E and F) Comparison of the residual KATP currents in the presence of 100 μM MgATP (E) and 1 mM MgATP (F) in the absence (open bars) and presence of 30 μM gliclazide (filled bars). Currents are expressed relative to those in the absence of both MgATP and gliclazide. The data were published previously by Proks et al. [25].
Mutation R201C is located in the putative ATP-binding site; it causes intermediate impairment of ATP inhibition (IC50=98 μM) [25] which results in permanent form of the disease [permanent ND (PNDM)], sometimes also associated with developmental delay (i-DEND syndrome) (intermediate DEND syndrome; ND with developmental delay and epilepsy); [40]. In the presence of 100 μM MgATP (Figure 3A, filled symbols), suppression of MgATP activation by gliclazide unmasks inhibitory effect of the nt which results in ∼50% reduction in the gliclazide-resistant current (since 100 μM is close to IC50 for the inhibitory effect of ATP). Note that there is also a small increase in IC50 for gliclazide block in the presence of MgATP attributable to displacement of the drug by the nt discussed above.
ATP-binding mutation G334D almost completely abolishes the inhibitory effect of ATP [31,32]; patients with this mutation have the most severe form of ND with developmental delay and epilepsy (DEND syndrome) [32]. Since the inhibitory effect of ATP is virtually absent from these channels, maximal extent of high-affinity sulphonylurea inhibition of KATP channels with this mutation is not enhanced by MgATP, even at a physiological concentration of the nt (1 mM; Figures 3C, 3E and 3F). In contrast, channels with mutation R201C are almost completely inhibited with 1 mM MgATP in the presence of the sulphonylurea (Figure 3F). This explains why sulphonylureas can be used to treat ND in patients with R201C but not G334D mutation [25,41].
Intermediate DEND syndrome mutation V59M [40] impairs gating of the channel and thus suppresses the direct inhibitory effect of gliclazide (Figures 1B and 3B, open circles). However, the maximal extent of the sulphonylurea inhibition is greatly enhanced in the presence of 100 μM MgATP as the drug unmasks substantial inhibitory effect of the nt at Kir6.2 (IC50 for the inhibitory effect of ATP is 62 μM) [25]. Thus, although inhibition by sulphonylurea alone is poor, these channels can be effectively blocked by the drug in the presence of physiological concentrations of ATP (1 mM, Figure 3F). This is not the case for mutation I296L, which is only partially blocked by gliclazide even in the presence of 3 mM of MgATP (Figure 3D). This mutation causes very strong gating defect that results in DEND syndrome; unlike for V59M, patients with this mutation cannot be treated with sulphonylureas [42].
In addition to the defects in ATP inhibition, some mutations in SUR cause ND by enhancing channel activation by Mg–nts [39]. Available data suggest that mutations discovered so far in this category may have either no effect or cause only small reduction in the indirect block of sulphonylureas [39], which is probably mediated via displacement of the drug from SUR1 by Mg–nt binding to NBS2 [43].
Concluding remarks
Effective inhibition of β-cell KATP channels by sulphonylureas requires co-operation from adenosine nts, which are major intracellular modulators of these channels. The same is true for glinides such as repaglinide [44]. Interestingly, recent data indicate that the collaboration between nts and sulphonylureas extends beyond the KATP channel. Drug-induced stimulation of insulin exocytosis via Epac2 (cAMP-activated guanine-nt exchange factor). It has recently been demonstrated that binding of sulphonylureas to this protein requires cAMP [45]. Understanding of the molecular mechanisms of action of sulphonylureas thus underlines the importance of studying drug effects within the context of the native cellular environment.
Acknowledgments
We would like to thank Sonja Fantom for technical help with preparing the figures.
Abbreviations
- ABC
ATP-binding cassette
- KATP
ATP-sensitive potassium channel
- NBD1/2
nt-binding domain 1/2
- NBS1/2
nt-binding site 1/2
- ND
neonatal diabetes
- nt
nucleotides
- PO
open probability
- SUR
sulphonylurea receptor
- TM
transmembrane domain
Footnotes
ATP binding cassette transporters: from mechanism to organism: Held at University of Chester, U.K., 16–18 April 2015.
Funding
HdW was supported by the European Foundation for the Study of Diabetes (EFSD). PP was supported by the Wellcome Trust.
References
- 1.Thulé P.M., Umpierrez G. Sulfonylureas: a new look at old therapy. Curr. Diab. Rep. 2014;14:473–481. doi: 10.1007/s11892-014-0473-5. [DOI] [PubMed] [Google Scholar]
- 2.Gribble F.M., Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia. 2003;46:875–891. doi: 10.1007/s00125-003-1143-3. [DOI] [PubMed] [Google Scholar]
- 3.Ashcroft F.M., Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog. Biophys. Mol. Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- 4.Proks P., Reimann F., Green N., Gribble F., Ashcroft F.M. Sulfonylurea stimulation of insulin secretion. Diabetes. 2002;51(Suppl 3):S368–S376. doi: 10.2337/diabetes.51.2007.S368. [DOI] [PubMed] [Google Scholar]
- 5.Renström E., Barg S., Thévenod F., Rorsman P. Sulfonylurea-mediated stimulation of insulin exocytosis via an ATP-sensitive K+ channel-independent action. Diabetes. 2002;51(Suppl 1):S33–S36. doi: 10.2337/diabetes.51.2007.S33. [DOI] [PubMed] [Google Scholar]
- 6.Zhang C.L., Katoh M., Shibasaki T., Minami K., Sunaga Y., Takahashi H., Yokoi N., Iwasaki M., Miki T., Seino S. The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science. 2009;325:607–610. doi: 10.1126/science.1172256. [DOI] [PubMed] [Google Scholar]
- 7.Ashcroft F.M. Adenosine 5'-triphosphate-sensitive potassium channels. Annu. Rev. Neurosci. 1988;11:97–118. doi: 10.1146/annurev.ne.11.030188.000525. [DOI] [PubMed] [Google Scholar]
- 8.Shyng S., Nichols C.G. Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tucker S.J., Gribble F.M., Zhao C., Trapp S., Ashcroft F.M. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- 10.Aittoniemi J., Fotinou C., Craig T.J., de Wet H., Proks P., Ashcroft F.M. SUR1: a unique ATP-binding cassette protein that functions as an ion channel regulator. Phil. Trans. Royal Soc. Lond. 2009;364:257–267. doi: 10.1098/rstb.2008.0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zingman L.V., Alekseev A.E., Bienengraeber M., Hodgson D., Karger A.B., Dzeja P.P., Terzic A. Signalling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron. 2001;31:233–245. doi: 10.1016/S0896-6273(01)00356-7. [DOI] [PubMed] [Google Scholar]
- 12.Li L., Geng X., Drain P. Open state destabilization by ATP occupancy is mechanism speeding burst exit underlying KATP channel inhibition by ATP. J. Gen. Physiol. 2002;119:105–116. doi: 10.1085/jgp.119.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarasov A.I., Girard C.A., Ashcroft F.M. ATP sensitivity of the ATP-sensitive K+ channel in intact and permeabilized pancreatic beta-cells. Diabetes. 2006;55:2446–2454. doi: 10.2337/db06-0360. [DOI] [PubMed] [Google Scholar]
- 14.Vergani P., Lockless S.W., Nairn A.C., Gadsby D.C. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 2005;433:876–80. doi: 10.1038/nature03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai M.F., Li M., Hwang T.C. Stable ATP binding mediated by a partial NBD dimer of the CFTR chloride channel. J. Gen. Physiol. 2010;135:399–414. doi: 10.1085/jgp.201010399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg M.F., Callaghan R., Ford R.C., Higgins C.F. Structure of the multidrug resistance P-glycoprotein to 2.5 nm resolution determined by electron microscopy and image analysis. J. Biol. Chem. 1997;272:10685–10694. doi: 10.1074/jbc.272.16.10685. [DOI] [PubMed] [Google Scholar]
- 17.Martin C., Berridge G., Mistry P., Higgins C., Charlton P., Callaghan R. Drug binding sites on P-glycoprotein are altered by ATP binding prior to nucleotide hydrolysis. Biochemistry. 2000;39:11901–11906. doi: 10.1021/bi000559b. [DOI] [PubMed] [Google Scholar]
- 18.Vila-Carriles W.H., Zhao G., Bryan J. Defining a binding pocket for sulphonylureas in ATP-sensitive potassium channels. FASEB J. 2007;21:18–25. doi: 10.1096/fj.06-6730hyp. [DOI] [PubMed] [Google Scholar]
- 19.Ashfield R., Gribble F.M., Ashcroft S.J., Ashcroft F.M. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the KATP channel. Diabetes. 1999;48:1341–1347. doi: 10.2337/diabetes.48.6.1341. [DOI] [PubMed] [Google Scholar]
- 20.Kühner P., Prager R., Stephan D., Russ U., Winkler M., Ortis D., Bryan J., Quast U. Importance of the Kir6.2 N-terminus for the interaction of glibenclamide and repaglinide with the pancreatic KATP channel. Naunyn-Schmiedeberg's Arch. Pharmacol. 2012;385:299–311. doi: 10.1007/s00210-011-0709-8. [DOI] [PubMed] [Google Scholar]
- 21.Hansen A.M.K., Hansen J.B., Carr R.D., Ashcroft F.M., Wahl P. Kir6.2-dependent high affinity repaglinide binding to beta-cell KATP channels. Br. J. Pharmacol. 2005;144:551–557. doi: 10.1038/sj.bjp.0706082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gribble F.M., Tucker S.J., Ashcroft F.M. The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. J. Physiol. 1997;504:35–45. doi: 10.1111/j.1469-7793.1997.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdelmoneim A.S., Hasenbank S.E., Seubert J.M., Brocks D.R., Light P.E., Simpson S.H. Variations in tissue selectivity amongst insulin secretagogues: a systematic review. Diabetes. Obes. Metab. 2012;14:130–138. doi: 10.1111/j.1463-1326.2011.01496.x. [DOI] [PubMed] [Google Scholar]
- 24.Proks P., de Wet H., Ashcroft F.M. Activation of the KATP channel by Mg-nucleotide interaction with SUR1. J. Gen. Physiol. 2010;136:389–405. doi: 10.1085/jgp.201010475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Proks P., de Wet H., Ashcroft F.M. Molecular mechanism of sulphonylurea block of KATP channels carrying mutations that impair ATP inhibition and cause neonatal diabetes. Diabetes. 2013;62:1–11. doi: 10.2337/db13-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trube G., Rorsman P., Ohno-Shosaku T. Opposite effects of tolbutamide and diazoxide on the ATP-dependent K- channel in mouse pancreatic beta cells. Pflügers Arch. 1986;407:493–499. doi: 10.1007/BF00657506. [DOI] [PubMed] [Google Scholar]
- 27.Lawrence C.L., Proks P., Rodrigo G.C., Jones P., Hayabuchi Y., Standen N.B., Ashcroft F.M. Gliclazide produces high-affinity block of KATP channels in mouse isolated pancreatic β-cells but not rat heart or arterial smooth muscle cells. Diabetologia. 2001;44:1019–1025. doi: 10.1007/s001250100595. [DOI] [PubMed] [Google Scholar]
- 28.Zünckler B.J., Lins S., Ohno-Shosaku T., Trube G., Panten U. Cytosolic ADP enhances the sensitivity of tolbutamide of ATP-dependent K+ channels from pancreatic β -cells. FEBS Lett. 1988;239:241–244. doi: 10.1016/0014-5793(88)80925-6. [DOI] [PubMed] [Google Scholar]
- 29.Schwanstecher C., Dickel C., Panten U. Cytosolic nucleotides enhance the tolbutamide sensitivity of the ATP-dependent K-channel in mouse pancreatic beta-cells by their combined actions at inhibitory and stimulatory sites. Mol. Pharmacol. 1992;41:480–486. [PubMed] [Google Scholar]
- 30.Proks P., de Wet H., Ashcroft F.M. Sulfonylureas suppress the stimulatory action of Mg-nucleotides on Kir6.2/SUR1 but not Kir6.2/SUR2A KATP channels: A mechanistic study. J. Gen. Physiol. 2014;144:469–486. doi: 10.1085/jgp.201411222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drain P., Li L.H., Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13953–13958. doi: 10.1073/pnas.95.23.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masia R., Koster J.C., Tumini S., Chiarelli F., Colombo C., Nichols C.G., Barbetti F. An ATP-binding mutation (G334D) in KCNJ11 is associated with a sulfonylurea-insensitive form of developmental delay, epilepsy, and neonatal diabetes. Diabetes. 2007;56:328–333. doi: 10.2337/db06-1275. [DOI] [PubMed] [Google Scholar]
- 33.de Wet H., Mikhailov M.V., Fotinou C., Dreger M., Craig T.J., Vénien-Bryan C., Ashcroft F.M. Studies of the ATPase activity of the ABC protein SUR1. FEBS J. 2007;274:3532–3544. doi: 10.1111/j.1742-4658.2007.05879.x. [DOI] [PubMed] [Google Scholar]
- 34.Hambrock A., Löffler-Walz C., Quast U. Glibenclamide binding to sulphonylurea receptor subtypes: dependence on adenine nucleotides. Br. J. Pharmacol. 2002;136:995–1004. doi: 10.1038/sj.bjp.0704801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venkatesh N., Lamp S.T., Weiss J.N. Sulfonylureas, ATP-sensitive K+ channels, and cellular K+ loss during hypoxia, ischemia, and metabolic inhibition in mammalian ventricle. Circ. Res. 1991;69:623–637. doi: 10.1161/01.RES.69.3.623. [DOI] [PubMed] [Google Scholar]
- 36.Ashcroft F.M. New uses for old drugs: neonatal diabetes and sulphonylureas. Cell Metab. 2010;11:179–181. doi: 10.1016/j.cmet.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 37.Proks P., Clark R. ATP-sensitive potassium channels in health and disease. In: Islam S., editor. Islets of Langerhans. Springer; 2015. pp. 305–336. [Google Scholar]
- 38.Koster J.C., Remedi M.S., Dao C., Nichols C.G. ATP and sulfonylurea sensitivity of mutant ATP-sensitive K+ channels in neonatal diabetes: implications for pharmacogenomic therapy. Diabetes. 2005;54:2645–2654. doi: 10.2337/diabetes.54.9.2645. [DOI] [PubMed] [Google Scholar]
- 39.Proks P. Neonatal diabetes caused by activating mutations in the sulphonylurea receptor. Diabetes Metab. J. 2013;37:157–164. doi: 10.4093/dmj.2013.37.3.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flanagan S.E., Edghill E.L., Gloyn A.L., Ellard S., Hattersley A.T. Mutations in KCNJ11, which encodes Kir6.2, are a common cause of diabetes diagnosed in the first 6 months of life, with the phenotype determined by genotype. Diabetologia. 2006;49:1190–1197. doi: 10.1007/s00125-006-0246-z. [DOI] [PubMed] [Google Scholar]
- 41.Drain P. ATP and sulfonylurea linkage in the KATP channel solves a diabetes puzzler. Diabetes. 2013;62:3666–3668. doi: 10.2337/db13-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Proks P., Girard C., Haider S., Gloyn A.L., Hattersley A.T., Sansom M.S., Ashcroft F.M. A gating mutation at the internal mouth of the Kir6.2 pore is associated with DEND syndrome. EMBO Rep. 2005;6:470–475. doi: 10.1038/sj.embor.7400393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ortiz D., Voyvodic P., Gossack L., Quast U., Bryan J. Two neonatal diabetes mutations on transmembrane helix 15 of SUR1 increase affinity for ATP and ADP at nucleotide binding domain 2. J. Biol. Chem. 2012;287:17985–17895. doi: 10.1074/jbc.M112.349019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dabrowski M., Wahl P., Holmes W.E., Ashcroft F.M. Effect of repaglinide on cloned beta cell, cardiac and smooth muscle types of ATP-sensitive potassium channels. Diabetologia. 2001;44:747–756. doi: 10.1007/s001250051684. [DOI] [PubMed] [Google Scholar]
- 45.Shibasaki T., Takahashi T., Takahashi H., Seino S. Cooperation between cAMP signalling and sulfonylurea in insulin secretion. Diabetes Obes. Metab. 2014;16(Suppl 1):S118–S125. doi: 10.1111/dom.12343. [DOI] [PubMed] [Google Scholar]